An Overview of Pharmacodynamic Modelling, Ligand-Binding Approach and Its Application in Clinical Practice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pharmacokinetics, Biodistribution, and Pharmacodynamics of Drug Delivery Systems
JPET Fast Forward. Published on March 5, 2019 as DOI: 10.1124/jpet.119.257113 This article has not been copyedited and formatted. The final version may differ from this version. JPET # 257113 Title: Pharmacokinetic and Pharmacodynamic Properties of Drug Delivery Systems Authors: Patrick M. Glassman, Vladimir R. Muzykantov Affiliation: Department of Systems Pharmacology and Translational Therapeutics, Perelman School of Medicine, University of Pennsylvania Address: 3400 Civic Center Boulevard, Bldg 421, Philadelphia, Pennsylvania 19104-5158, United States Downloaded from jpet.aspetjournals.org at ASPET Journals on September 24, 2021 1 JPET Fast Forward. Published on March 5, 2019 as DOI: 10.1124/jpet.119.257113 This article has not been copyedited and formatted. The final version may differ from this version. JPET # 257113 Running Title: PK/PD Properties of Drug Delivery Systems Corresponding Authors: Vladimir R. Muzykantov ([email protected], (215) 898-9823) and Patrick M. Glassman ([email protected]) # of Text Pages: 22 # of Tables: 2 # of Figures: 4 Word Count – Abstract: 144 Word Count – Introduction: 350 Word Count – Discussion: N/A Downloaded from Non-Standard Abbreviations: Absorption, Distribution, Metabolism, and Elimination (ADME) Biodistribution (BD) Drug Delivery Systems (DDSs) Enhanced Permeability & Retention (EPR) jpet.aspetjournals.org Gastrointestinal (GI) Intravenously (IV) Neonatal Fc Receptor (FcRn) Monoclonal Antibody (mAb) Reticuloendothelial System (RES) at ASPET Journals on September 24, 2021 Pharmacodynamics (PD) Pharmacokinetics (PK) Physiologically-Based Pharmacokinetic (PBPK) Subcutaneously (SC) Target-Mediated Drug Disposition (TMDD) Recommended Section: Drug Discovery and Translational Medicine 2 JPET Fast Forward. Published on March 5, 2019 as DOI: 10.1124/jpet.119.257113 This article has not been copyedited and formatted. -

Clinical Pharmacology 1: Phase 1 Studies and Early Drug Development
Clinical Pharmacology 1: Phase 1 Studies and Early Drug Development Gerlie Gieser, Ph.D. Office of Clinical Pharmacology, Div. IV Objectives • Outline the Phase 1 studies conducted to characterize the Clinical Pharmacology of a drug; describe important design elements of and the information gained from these studies. • List the Clinical Pharmacology characteristics of an Ideal Drug • Describe how the Clinical Pharmacology information from Phase 1 can help design Phase 2/3 trials • Discuss the timing of Clinical Pharmacology studies during drug development, and provide examples of how the information generated could impact the overall clinical development plan and product labeling. Phase 1 of Drug Development CLINICAL DEVELOPMENT RESEARCH PRE POST AND CLINICAL APPROVAL 1 DISCOVERY DEVELOPMENT 2 3 PHASE e e e s s s a a a h h h P P P Clinical Pharmacology Studies Initial IND (first in human) NDA/BLA SUBMISSION Phase 1 – studies designed mainly to investigate the safety/tolerability (if possible, identify MTD), pharmacokinetics and pharmacodynamics of an investigational drug in humans Clinical Pharmacology • Study of the Pharmacokinetics (PK) and Pharmacodynamics (PD) of the drug in humans – PK: what the body does to the drug (Absorption, Distribution, Metabolism, Excretion) – PD: what the drug does to the body • PK and PD profiles of the drug are influenced by physicochemical properties of the drug, product/formulation, administration route, patient’s intrinsic and extrinsic factors (e.g., organ dysfunction, diseases, concomitant medications, -

Principle of Pharmacodynamics
Principle of pharmacodynamics Dr. M. Emamghoreishi Full Professor Department of Pharmacology Medical School Shiraz University of Medical Sciences Email:[email protected] Reference: Basic & Clinical Pharmacology: Bertrum G. Katzung and Anthony J. Treveror, 13th edition, 2015, chapter 20, p. 336-351 Learning Objectives: At the end of sessions, students should be able to: 1. Define pharmacology and explain its importance for a clinician. 2. Define ―drug receptor‖. 3. Explain the nature of drug receptors. 4. Describe other sites of drug actions. 5. Explain the drug-receptor interaction. 6. Define the terms ―affinity‖, ―intrinsic activity‖ and ―Kd‖. 7. Explain the terms ―agonist‖ and ―antagonist‖ and their different types. 8. Explain chemical and physiological antagonists. 9. Explain the differences in drug responsiveness. 10. Explain tolerance, tachyphylaxis, and overshoot. 11. Define different dose-response curves. 12. Explain the information that can be obtained from a graded dose-response curve. 13. Describe the potency and efficacy of drugs. 14. Explain shift of dose-response curves in the presence of competitive and irreversible antagonists and its importance in clinical application of antagonists. 15. Explain the information that can be obtained from a quantal dose-response curve. 16. Define the terms ED50, TD50, LD50, therapeutic index and certain safety factor. What is Pharmacology?It is defined as the study of drugs (substances used to prevent, diagnose, and treat disease). Pharmacology is the science that deals with the interactions betweena drug and the bodyor living systems. The interactions between a drug and the body are conveniently divided into two classes. The actions of the drug on the body are termed pharmacodynamicprocesses.These properties determine the group in which the drug is classified, and they play the major role in deciding whether that group is appropriate therapy for a particular symptom or disease. -

In Silico Pharmacodynamics, Toxicity Profile and Biological Activities of the Saharan Medicinal Plant Limoniastrum Feei
Brazilian Journal of Pharmaceutical Sciences Article http://dx.doi.org/10.1590/s2175-97902017000300061 In silico pharmacodynamics, toxicity profile and biological activities of the Saharan medicinal plant Limoniastrum feei Ouahab Ammar* Department of Pharmacy, Faculty of Medical Sciences, University of Batna 2, Algeria In-silico study was performed to find the pharmacodynamics, toxicity profiles and biological activities of three phytochemicals isolated from Limoniastrum feei (Plumbagenaceae). Online pharmacokinetic tools were used to estimate the potential of Quercetin, kaempferol-3-O-β-D-glucopyranoside (astragalin) and quercitin-7-O-β-D-glucopyranoside as specific drugs. Then the prediction of potential targets of these compounds were investigated using PharmMapper. Auto-Dock 4.0 software was used to investigate the different interactions of these compounds with the targets predicted earlier. The permeability of quercetin s rule of five. Hematopoietic prostaglandin (PG) D׳ was found within the range stated by Lipinski synthase (HPGDS), farnesyl diphosphate synthetase (FPPS) and the deoxycytidine kinase (DCK) were potential targets for quercetin, astragalin and quercetin 7, respectively. Quercetin showed antiallergic and anti-inflammatory activity, while astragalin and quercetin 7 were predicted to have anticancer activities. The activity of Astragalin appeared to be mediated by FPPS inhibition. The inhibition of DCK was predicted as the anticancer mechanisms of quercetin 7. The compounds showed interesting interactions and satisfactory binding energies when docked into their targets. These compounds are proposed to have activities against a variety of human aliments such as allergy, tumors, muscular dystrophy, and diabetic cataracts. Keywords: Limoniastrum feei/pharmacokinetics. Limoniastrum feei/biological activity. Quercetin. Astragalin. Quercetin 7. Medicinal plants. Molecular docking. -

Pharmacology Part 2: Introduction to Pharmacokinetics
J of Nuclear Medicine Technology, first published online May 3, 2018 as doi:10.2967/jnmt.117.199638 PHARMACOLOGY PART 2: INTRODUCTION TO PHARMACOKINETICS. Geoffrey M Currie Faculty of Science, Charles Sturt University, Wagga Wagga, Australia. Regis University, Boston, USA. Correspondence: Geoff Currie Faculty of Science Locked Bag 588 Charles Sturt University Wagga Wagga 2678 Australia Telephone: 02 69332822 Facsimile: 02 69332588 Email: [email protected] Foot line: Introduction to Pharmacokinetics 1 Abstract Pharmacology principles provide key understanding that underpins the clinical and research roles of nuclear medicine practitioners. This article is the second in a series of articles that aims to enhance the understanding of pharmacological principles relevant to nuclear medicine. This article will build on the introductory concepts, terminology and principles of pharmacodynamics explored in the first article in the series. Specifically, this article will focus on the basic principles associated with pharmacokinetics. Article 3 will outline pharmacology relevant to pharmaceutical interventions and adjunctive medications employed in general nuclear medicine, the fourth pharmacology relevant to pharmaceutical interventions and adjunctive medications employed in nuclear cardiology, the fifth the pharmacology related to contrast media associated with computed tomography (CT) and magnetic resonance imaging (MRI), and the final article will address drugs in the emergency trolley. 2 Introduction As previously outlined (1), pharmacology is the scientific study of the action and effects of drugs on living systems and the interaction of drugs with living systems (1-7). For general purposes, pharmacology is divided into pharmacodynamics and pharmacokinetics (Figure 1). The principle of pharmacokinetics is captured by philosophy of Paracelsus (medieval alchemist); “only the dose makes a thing not a poison” (1,8,9). -

Pharmacokinetics and Pharmacology of Drugs Used in Children
Drug and Fluid Th erapy SECTION II Pharmacokinetics and Pharmacology of Drugs Used CHAPTER 6 in Children Charles J. Coté, Jerrold Lerman, Robert M. Ward, Ralph A. Lugo, and Nishan Goudsouzian Drug Distribution Propofol Protein Binding Ketamine Body Composition Etomidate Metabolism and Excretion Muscle Relaxants Hepatic Blood Flow Succinylcholine Renal Excretion Intermediate-Acting Nondepolarizing Relaxants Pharmacokinetic Principles and Calculations Atracurium First-Order Kinetics Cisatracurium Half-Life Vecuronium First-Order Single-Compartment Kinetics Rocuronium First-Order Multiple-Compartment Kinetics Clinical Implications When Using Short- and Zero-Order Kinetics Intermediate-Acting Relaxants Apparent Volume of Distribution Long-Acting Nondepolarizing Relaxants Repetitive Dosing and Drug Accumulation Pancuronium Steady State Antagonism of Muscle Relaxants Loading Dose General Principles Central Nervous System Effects Suggamadex The Drug Approval Process, the Package Insert, and Relaxants in Special Situations Drug Labeling Opioids Inhalation Anesthetic Agents Morphine Physicochemical Properties Meperidine Pharmacokinetics of Inhaled Anesthetics Hydromorphone Pharmacodynamics of Inhaled Anesthetics Oxycodone Clinical Effects Methadone Nitrous Oxide Fentanyl Environmental Impact Alfentanil Oxygen Sufentanil Intravenous Anesthetic Agents Remifentanil Barbiturates Butorphanol and Nalbuphine 89 A Practice of Anesthesia for Infants and Children Codeine Antiemetics Tramadol Metoclopramide Nonsteroidal Anti-infl ammatory Agents 5-Hydroxytryptamine -

White Paper Recommendations for the Development and Validation Of
The AAPS Journal ( # 2017) DOI: 10.1208/s12248-017-0181-6 White Paper Recommendations for the Development and Validation of Neutralizing Antibody Assays in Support of Biosimilar Assessment D. Gouty,1,16 C. C. Cai,2 X. Y. Cai,3 A. Kasinath,4 V. Kumar,5 S. Alvandkouhi,6 J. Yang,7 S. Pederson,8 B. Babbitt,2 D. Peritt,9 A. Rudy,10 V. Koppenburg,11 A. Dasilva,12 M. Ullmann,13 S. Liu,14 and C. Satterwhite15 Received 23 May 2017; accepted 21 November 2017 Abstract. The American Association of Pharmaceutical Scientists (AAPS) biosimilar focus group on nonclinical and clinical assays has developed this manuscript to guide the industry on best practices and testing strategies when developing neutralizing antibody (NAb) assays for biosimilar programs. The immunogenicity assessment to biosimilar and originator drug products is one of the key aspects of clinical programs for biosimilars to demonstrate biosimilarity. Establishing that there are no clinically meaningful differences in immune response between a proposed product and the originator product is a key element in the demonstration of biosimilarity. It is critical to collect, evaluate, and compare the safety and immunogenicity data from the clinical pharmacology, safety, and/or efficacy studies especially when the originator drug product is known to have potential for immune-mediated toxicity. This manuscript aims to provide a comprehensive review and recommendations on assay formats, critical reagents, approaches to method development, and validation of the neutralizing antibody assays in extrapolation within the scope of biosimilar drug development programs. Even if there are multiple options on the development and validation of NAb assays for biosimilar programs, the type of drug and its MoA will help determine the assay format and technical platform for NAb assessment (e.g., cell-based or non-cell-based assay). -

International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification
0031-6997/03/5504-597–606$7.00 PHARMACOLOGICAL REVIEWS Vol. 55, No. 4 Copyright © 2003 by The American Society for Pharmacology and Experimental Therapeutics 30404/1114803 Pharmacol Rev 55:597–606, 2003 Printed in U.S.A International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology RICHARD R. NEUBIG, MICHAEL SPEDDING, TERRY KENAKIN, AND ARTHUR CHRISTOPOULOS Department of Pharmacology, University of Michigan, Ann Arbor, Michigan (R.R.N.); Institute de Recherches Internationales Servier, Neuilly sur Seine, France (M.S.); Systems Research, GlaxoSmithKline Research and Development, Research Triangle Park, North Carolina (T.K.); and Department of Pharmacology, University of Melbourne, Parkville, Australia (A.C.) Abstract ............................................................................... 597 I. Introduction............................................................................ 597 II. Working definition of a receptor .......................................................... 598 III. Use of drugs in definition of receptors or of signaling pathways ............................. 598 A. The expression of amount of drug: concentration and dose ............................... 598 1. Concentration..................................................................... 598 2. Dose. ............................................................................ 598 B. General terms used to describe drug action ........................................... -

Primer on Forensic Toxicology
1 OSAC’s purpose is to strengthen the nation's use of forensic science by providing technical leadership necessary to facilitate the development and promulgation of consensus-based documentary standards and guidelines for forensic science, promoting standards and guidelines that are fit-for-purpose and based on sound scientific principles, promoting the use of OSAC standards and guidelines by accreditation and certification bodies, and establishing and maintaining working relationships with other similar organizations. 2 The six subcommittees of the Chemistry/Instrumental Analysis SAC include Fire Debris and Explosives, Geological Materials, Gunshot Residue, Materials (Trace), Seized Drugs and Toxicology. 3 This resource is intended to provide a brief overview, or the “what”, “why”, “where”, and “how”, of the field of Forensic Toxicology. 4 Toxicology is the study of drugs and chemicals on biological systems. Toxicology relies on information from numerous scientific and social disciplines as well as engineering and statistics. 5 Forensic toxicology deals with the application of toxicology to cases and issues where adverse effects of the use of impairing, toxic and lethal concentrations of drugs have administrative or medicolegal consequences, and where the results are likely to be used in a legal setting. 6 Other career opportunities exist in hospitals, universities, and industry. 7 Paracelsus is often credited with one of the original conceivers of toxicology. Manly Hall, a famous author of the 19th Century called him "the precursor of chemical pharmacology and therapeutics and the most original medical thinker of the sixteenth century.“ A principle tenet of forensic toxicology is that the “dose makes something a poison”. For example, water is generally a safe and essential beverage all living organisms require to live, but if taken in an excessive amount this can interfere with a body’s chemistry and create a toxic environment. -

Objectives Pharmacodynamics Las Pharmadynamics (Cont)
Pharmacodynamics Locals have 3 basic components Amine group Aromatic group Linkage group Adding carbons = lipophilicity, duration of Brian D. Berry Jr., CRNA, MS action, and protein binding Adjunct Faculty Reversibly block University of Pittsburgh conduction of impulses along central and peripheral nerve pathways UPMC Staffing Network, Nurse Anesthesia (autonomic, somatic University of Pittsburgh Medical Center sensory and somatic motor) Department of Anesthesia 1 4 LAs pharmadynamics (cont) Weak bases Increased affinity for αasubunit open Na+ channels and 75% of channels must be β subunit blocked for effect H type alpha subunit binds to local anesthetics 2 5 Objectives Discuss and Review the pharmacodynamics/pharmacokenetics of local anesthetics H α - Subunit Differentiate between local anesthetics and their potential to cause local toxicity Discuss the mechanism of action of local toxicity Discuss current literature regarding lipid emulsion therapy 3 6 Na+ molecule LA LA+ 7 10 Pharmacokinetics Toxicity Absorption, Distribution, Metabolism, Good News…incidence has decreased and Excretion 7.5-20 per 10,000 peripheral nerve blocks 4 per 10,000 epidurals Absorption influenced by site, dose, and use of epi Redistribution from vessel rich group to Toxicity most often to intravascular vessel poor group injection and not accumulation Amide LAs metabolized by CYP3A4. Ester broken down my plasma Saftey has d/t ○ Aspiration cholinesterase ○ Knowledge (i.e. local toxic doses) Renal excretion ○ Divided doses ○ Test dose -

International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology
0031-6997/03/5504-597–606$7.00 PHARMACOLOGICAL REVIEWS Vol. 55, No. 4 Copyright © 2003 by The American Society for Pharmacology and Experimental Therapeutics 30404/1114803 Pharmacol Rev 55:597–606, 2003 Printed in U.S.A International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology RICHARD R. NEUBIG, MICHAEL SPEDDING, TERRY KENAKIN, AND ARTHUR CHRISTOPOULOS Department of Pharmacology, University of Michigan, Ann Arbor, Michigan (R.R.N.); Institute de Recherches Internationales Servier, Neuilly sur Seine, France (M.S.); Systems Research, GlaxoSmithKline Research and Development, Research Triangle Park, North Carolina (T.K.); and Department of Pharmacology, University of Melbourne, Parkville, Australia (A.C.) Abstract ............................................................................... 597 I. Introduction............................................................................ 597 II. Working definition of a receptor .......................................................... 598 III. Use of drugs in definition of receptors or of signaling pathways ............................. 598 A. The expression of amount of drug: concentration and dose ............................... 598 Downloaded from 1. Concentration..................................................................... 598 2. Dose. ............................................................................ 598 B. General terms used to describe drug action ........................................... -
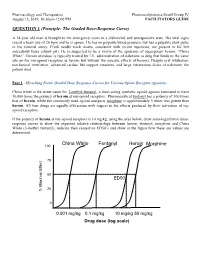
QUESTION 1 (Principle: the Graded Dose-Response Curve)
Pharmacology and Therapeutics Pharmacodynamics Small Group IV August 15, 2019, 10:30am-12:00 PM FACILITATORS GUIDE QUESTION 1 (Principle: The Graded Dose-Response Curve) A 34-year old man is brought to the emergency room in a disheveled and unresponsive state. His vital signs reveal a heart rate of 26 bpm and he is apneic. He has no palpable blood pressure, but has a palpable slow pulse in his femoral artery. Fresh needle track marks, consistent with recent injections, are present in his left antecubital fossa (elbow pit). He is suspected to be a victim of the epidemic of superpotent heroin, “China White”. Heroin overdose is typically treated by I.V. administration of naloxone (a drug that binds to the same site on the mu-opioid receptors as heroin, but without the narcotic effects of heroin). Despite oral intubation, mechanical ventilation, advanced cardiac life support measures, and large intravenous doses of naloxone, the patient died. Part 1 (Teaching Point: Graded Dose Response Curves for Various Opiate Receptor Agonists) China white is the street name for 3-methyl-fentanyl, a short-acting synthetic opioid agonist estimated to have 10,000 times the potency of heroin at mu-opioid receptors. Pharmaceutical fentanyl has a potency of 100 times that of heroin, while the commonly used opioid analgesic morphine is approximately 5 times less potent than heroin. All four drugs are equally efficacious with respect to the effects produced by their activation of m opioid receptors. If the potency of heroin at mu-opioid receptors is 10 mg/kg, using the axes below, draw semi-logarithmic dose- response curves to show the expected relative relationships between heroin, fentanyl, morphine and China White (3-methyl fentanyl), indicate their respective ED50’s and show in the figure how these are values are determined.