NMR Chemical Shift Analysis Decodes Olefin Oligo- and Polymerization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Graphene Supported Rhodium Nanoparticles for Enhanced Electrocatalytic Hydrogen Evolution Reaction Ameerunisha Begum1*, Moumita Bose2 & Golam Moula2
www.nature.com/scientificreports OPEN Graphene Supported Rhodium Nanoparticles for Enhanced Electrocatalytic Hydrogen Evolution Reaction Ameerunisha Begum1*, Moumita Bose2 & Golam Moula2 Current research on catalysts for proton exchange membrane fuel cells (PEMFC) is based on obtaining higher catalytic activity than platinum particle catalysts on porous carbon. In search of a more sustainable catalyst other than platinum for the catalytic conversion of water to hydrogen gas, a series of nanoparticles of transition metals viz., Rh, Co, Fe, Pt and their composites with functionalized graphene such as RhNPs@f-graphene, CoNPs@f-graphene, PtNPs@f-graphene were synthesized and characterized by SEM and TEM techniques. The SEM analysis indicates that the texture of RhNPs@f- graphene resemble the dispersion of water droplets on lotus leaf. TEM analysis indicates that RhNPs of <10 nm diameter are dispersed on the surface of f-graphene. The air-stable NPs and nanocomposites were used as electrocatalyts for conversion of acidic water to hydrogen gas. The composite RhNPs@f- graphene catalyses hydrogen gas evolution from water containing p-toluene sulphonic acid (p-TsOH) at an onset reduction potential, Ep, −0.117 V which is less than that of PtNPs@f-graphene (Ep, −0.380 V) under identical experimental conditions whereas the onset potential of CoNPs@f-graphene was at Ep, −0.97 V and the FeNPs@f-graphene displayed onset potential at Ep, −1.58 V. The pure rhodium nanoparticles, RhNPs also electrocatalyse at Ep, −0.186 V compared with that of PtNPs at Ep, −0.36 V and that of CoNPs at Ep, −0.98 V. -
![Anagostic Interactions Under Pressure: Attractive Or Repulsive? Wolfgang Scherer *[A], Andrew C](https://docslib.b-cdn.net/cover/5995/anagostic-interactions-under-pressure-attractive-or-repulsive-wolfgang-scherer-a-andrew-c-885995.webp)
Anagostic Interactions Under Pressure: Attractive Or Repulsive? Wolfgang Scherer *[A], Andrew C
Manuscript Click here to download Manuscript: manuscript_final.pdf ((Attractive or repulsive?)) DOI: 10.1002/anie.200((will be filled in by the editorial staff)) Anagostic Interactions under Pressure: Attractive or Repulsive? Wolfgang Scherer *[a], Andrew C. Dunbar [a], José E. Barquera-Lozada [a], Dominik Schmitz [a], Georg Eickerling [a], Daniel Kratzert [b], Dietmar Stalke [b], Arianna Lanza [c,d], Piero Macchi*[c], Nicola P. M. Casati [d], Jihaan Ebad-Allah [a] and Christine Kuntscher [a] The term “anagostic interactions” was coined in 1990 by Lippard preagostic interactions are considered to lack any “involvement of 2 and coworkers to distinguish sterically enforced M•••H-C contacts dz orbitals in M•••H-C interactions” and rely mainly on M(dxz, yz) (M = Pd, Pt) in square-planar transition metal d8 complexes from o V (C-H) S-back donation.[3b] attractive, agostic interactions.[1a] This classification raised the fundamental question whether axial M•••H-C interaction in planar The first observation of unusual axial M•••H-C interaction in 8 8 d -ML4 complexes represent (i) repulsive anagostic 3c-4e M•••H-C planar d -ML4 complexes was made by S. Trofimenko, who interactions[1] (Scheme 1a) or (ii) attractive 3c-4e M•••H-C pioneered the chemistry of transition metal pyrazolylborato hydrogen bonds[2] (Scheme 1b) in which the transition metal plays complexes.[5,6] Trofimenko also realized in 1968, on the basis of the role of a hydrogen-bond acceptor (Scheme 1b). The latter NMR studies, that the shift of the pseudo axial methylene protons in 3 bonding description is related to another bonding concept which the agostic species [Mo{Et2B(pz)2}(K -allyl)(CO)2] (1) (pz = describes these M•••H-C contacts in terms of (iii) pregostic or pyrazolyl; allyl = H2CCHCH2) “is comparable in magnitude but [3] preagostic interactions (Scheme 1c) which are considered as being different in direction from that observed in Ni[Et2B(pz)2]2” (2) “on the way to becoming agostic, or agostic of the weak type”.[4] (Scheme 2).[6,7] Scheme 1. -

Alkyne Metathesis Catalysts: Scope and Future André Mortreux, Olivier Coutelier
Alkyne Metathesis Catalysts: Scope And Future André Mortreux, Olivier Coutelier To cite this version: André Mortreux, Olivier Coutelier. Alkyne Metathesis Catalysts: Scope And Future. Journal of Molecular Catalysis A: Chemical, Elsevier, 2006, 254, pp.96-104. 10.1016/j.molcata.2006.03.054. hal-00107451 HAL Id: hal-00107451 https://hal.archives-ouvertes.fr/hal-00107451 Submitted on 18 Oct 2006 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ALKYNE METATHESIS CATALYSTS: SCOPE AND FUTURE. André Mortreux*, Olivier Coutelier Laboratoire de Catalyse de Lille UMR 8010 CNRS USTL-ENSCL, BP 90108, 59652 Villeneuve d’Ascq Cedex France Corresponding author.Tel +33320434993 ;Fax+33320434486 E-mail adress: [email protected] ABSTRACT This paper presents the evolution of alkyne metathesis since the early discoveries, essentially from the catalyst point of view. It is shown that although well defined carbynes may be useful for this reaction, further work has been made, aimed at the synthesis of new catalysts or catalytic systems, based on molybdenum precursors , associated or not with phenolic co- catalysts. The major objectives have been to obtain more functional groups tolerants catalysts, for their application in organic synthesis, including RCM for further stereoselective hydrogenation of the triple bond in the cycle, as well as for polymerization of aromatic diynes. -

Organometallics Study Meeting H.Mitsunuma 1
04/21/2011 Organometallics Study Meeting H.Mitsunuma 1. Crystal field theory (CFT) and ligand field theory (LFT) CFT: interaction between positively charged metal cation and negative charge on the non-bonding electrons of the ligand LFT: molecular orbital theory (back donation...etc) Octahedral (figure 9-1a) d-electrons closer to the ligands will have a higher energy than those further away which results in the d-orbitals splitting in energy. ligand field splitting parameter ( 0): energy between eg orbital and t2g orbital 1) high oxidation state 2) 3d<4d<5d 3) spectrochemical series I-< Br-< S2-< SCN-< Cl-< N -,F-< (H N) CO, OH-< ox, O2-< H O< NCS- <C H N, NH < H NCH CH NH < bpy, phen< NO - - - - 3 2 2 2 5 5 3 2 2 2 2 2 < CH3 ,C6H5 < CN <CO cf) pairing energy: energy cost of placing an electron into an already singly occupied orbital Low spin: If 0 is large, then the lower energy orbitals(t2g) are completely filled before population of the higher orbitals(eg) High spin: If 0 is small enough then it is easier to put electrons into the higher energy orbitals than it is to put two into the same low-energy orbital, because of the repulsion resulting from matching two electrons in the same orbital 3 n ex) (t2g) (eg) (n= 1,2) Tetrahedral (figure 9-1b), Square planar (figure 9-1c) LFT (figure 9-3, 9-4) - - Cl , Br : lower 0 (figure 9-4 a) CO: higher 0 (figure 9-4 b) 2. Ligand metal complex hapticity formal chargeelectron donation metal complex hapticity formal chargeelectron donation MR alkyl 1 -1 2 6-arene 6 0 6 MH hydride 1 -1 2 M MX H 1 -1 2 halogen 1 -1 2 M M -hydride M OR alkoxide 1 -1 2 X 1 -1 4 M M -halogen O R acyl 1 -1 2 O 1 -1 4 M R M M -alkoxide O 1-alkenyl 1 -1 2 C -carbonyl 1 0 2 M M M R2 C -alkylidene 1 -2 4 1-allyl 1 -1 2 M M M O C 3-carbonyl 1 0 2 M R acetylide 1 -1 2 MMM R R C 3-alkylidine 1 -3 6 M carbene 1 0 2 MMM R R M carbene 1 -2 4 R M carbyne 1 -3 6 M CO carbonyl 1 0 2 M 2-alkene 2 0 2 M 2-alkyne 2 0 2 M 3-allyl 3 -1 4 M 4-diene 4 0 4 5 -cyclo 5 -1 6 M pentadienyl 1 3. -

Agostic Interaction and Intramolecular Proton Transfer from the Protonation
Agostic interaction and intramolecular proton SPECIAL FEATURE transfer from the protonation of dihydrogen ortho metalated ruthenium complexes Andrew Toner†, Jochen Matthes†‡, Stephan Gru¨ ndemann‡, Hans-Heinrich Limbach‡, Bruno Chaudret†, Eric Clot§, and Sylviane Sabo-Etienne†¶ †Laboratoire de Chimie de Coordination du Centre National de la Recherche Scientifique, Associe´a` l’Universite´Paul Sabatier, 205 route de Narbonne, 31077 Toulouse Cedex 04, France; ‡Institute of Chemistry, Freie Universita¨t Berlin, Takustrasse 3, D-14195 Berlin, Germany; and §Laboratoire de Structure et Dynamique des Syste`mes Mole´culaires et Solides (Centre National de la Recherche Scientifique, Unite´Mixte de Recherche 5253), Institut Charles Gerhardt, case courrier 14, Universite´Montpellier II, Place Euge`ne Bataillon, 34000 Montpellier, France Edited by Jay A. Labinger, California Institute of Technology, Pasadena, CA, and accepted by the Editorial Board January 17, 2007 (received for review October 13, 2006) Protonation of the ortho-metalated ruthenium complexes insertion of an olefin into the aromatic C–H bond ortho to an i phenylpyridine (ph-py) (1), benzoquinoline activating ketone group (Eq. 1) (30). In this system, the key-2 ؍ RuH(H2)(X)(P Pr3)2 [X i ؉ ؊ bq) (2)] and RuH(CO)(ph-py)(P Pr3)2 (3) with [H(OEt2)2] [BAr4] intermediate is an ortho-metalated complex resulting from ortho) CF3)2C6H3)4B]) under H2 atmosphere yields the corre- C–H bond activation, thanks to chelating assistance with the donor)-3,5)] ؍ BAr4) sponding cationic hydrido dihydrogen ruthenium complexes group. Coordination of the olefin, olefin insertion into Ru–H and i phenylpyridine (ph-py) (1-H); ben- C–C coupling are the subsequent steps needed to close the catalytic ؍RuH(H2)(H-X)(P Pr3)2][BAr4][X] zoquinoline (bq) (2-H)] and the carbonyl complex [RuH(CO)(H-ph- cycle as proposed by Kakiuchi and Murai (8). -

20 Catalysis and Organometallic Chemistry of Monometallic Species
20 Catalysis and organometallic chemistry of monometallic species Richard E. Douthwaite Department of Chemistry, University of York, Heslington, York, UK YO10 5DD Organometallic chemistry reported in 2003 again demonstrated the breadth of interest and application of this core chemical field. Significant discoveries and developments were reported particularly in the application and understanding of organometallic compounds in catalysis. Highlights include the continued develop- ment of catalytic reactions incorporating C–H activation processes, the demonstra- 1 tion of inverted electronic dependence in ligand substitution of palladium(0), and the synthesis of the first early-transition metal perfluoroalkyl complexes.2 1 Introduction A number of relevant reviews and collections of research papers spanning the transition metal series were published in 2003. The 50th anniversary of Ziegler catalysis was commemorated3,4 and a survey of metal mediated polymerisation using non-metallocene catalysts surveyed.5 Journal issues dedicated to selected topics included metal–carbon multiple bonds and related organometallics,6 developments in the reactivity of metal allyl and alkyl complexes,7 metal alkynyls,8 and carbon rich organometallic compounds including 1.9 Reviews of catalytic reactions using well-defined precatalyst complexes include alkene ring-closing and opening methathesis using molybdenum and tungsten imido DOI: 10.1039/b311797a Annu. Rep. Prog. Chem., Sect. A, 2004, 100, 385–406 385 alkylidene precatalysts,10 chiral organometallic half-sandwich -

Characterization of Agostic Interactions in Theory and Computation
Characterization of agostic interactions in theory and computation Matthias Lein Centre for Theoretical Chemistry and Physics, New Zealand Institute for Advanced Study, Massey University Auckland Abstract Agostic interactions are covalent intramolecular interactions between an electron deficient metal and a σ-bond in close geometrical proximity to the metal atom. While the classic cases involve CH σ-bonds close to early transition metals like ti- tanium, many more agostic systems have been proposed which contain CH, SiH, BH, CC and SiC σ-bonds coordinated to a wide range of metal atoms. Recent computational studies of a multitude of agostic interactions are reviewed in this contribution. It is highlighted how several difficulties with the theoretical descrip- tion of the phenomenon arise because of the relative weakness of this interaction. The methodology used to compute and interpret agostic interactions is presented and different approaches such as atoms in molecules (AIM), natural bonding orbitals (NBO) or the electron localization function (ELF) are compared and put into con- text. A brief overview of the history and terminology of agostic interactions is given in the introduction and fundamental differences between α, β and other agostic interactions are explained. Key words: agostic interactions, chemical bond, density functional method, ab-initio calculations, computational chemistry, natural bond orbitals, atoms in molecules, electron localization function Contents 1 Introduction 2 arXiv:0807.1751v1 [physics.chem-ph] 10 Jul 2008 1.1 Agostic Interactions by type 3 2 Agostic Interactions by method 6 2.1 Computational Approach 7 2.2 Spectroscopic Approach 16 3 Conclusions 18 4 Acknowledgments 19 References 19 Preprint submitted to Elsevier 22 October 2018 1 Introduction Transition metal compounds which exhibit a close proximity of CH systems to the metal atom were discovered relatively early in the mid 1960’s and early 1970’s as the quality and availability of x-ray crystallography improved [1,2,3,4,5,6]. -
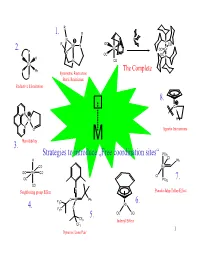
Strategies to Introduce „Free Coordination Sites“ 1. 2. 3. 4. 5. 6. 7. 8
R 1. R N P R OC P Mo N CO 2. Cr OC Cr P N OC OC P H P N CO Ru TheSteric Complete Restriction P Ph Symmetric Restriction P Steric Restriction Reductive Elimination 8. Co N Pd H N O Agostic Interactions Hemilability M 3. Strategies to introduce „Free coordination sites“ PCy3 Cl H Ph CO Ru OC Mn CO Cl OC PCy3 7. CO Neighboring group Effect N Pseudo-Jahn-Teller-Effect O W Ph F C 3 Rh 6. 4. O F3C 5. OC CO CF3 Indenyl Effect CF3 1 Dynamic Lone Pair 1. Steric restrictions + - N2 + 6 H + 6e R R R R R R R R cat. N N N R R R R N 2 NH3 N N Mo N Mo N + - N N H , e N N R R + - R R -NH3 H , e NH R R R R R R N N Mo N R N R N + - H2N H , e N Mo N N N R R R R R R R R R R R R + - NH2 H , e R R R R H+, e- R + - N R N H , e HN N N Mo N N Mo N -NH3 Mo N N N N N N N 2 Symmetric restrictions R R 1. R R + - N N2 + 6 H + 6e N R cat. R N Mo N 2 NH3 N N 15e-complex rather than 17e-complex E one lone pair is non-bonding! 3 Electron-transfer-Catalysis 1. Electronic restrictions CH3 CH3 PPh 18e-Complex 3 18e-Complex Mn Mn CO CO H CCN slow Ph P 3 CO 3 CO o E =0.19V Eo=0.52V CH3 CH3 PPh 3 17e-Complex 17e-Complex Mn fast Mn CO CO H CCN Ph P 3 CO 3 CO CH3 Mn CO H3CCN CO The reaction works catalytically, because PPh 3 is a stronger π-acid than CH 3CN. -

Symmetry 2010, 2, 1745-1762; Doi:10.3390/Sym2041745 OPEN ACCESS Symmetry ISSN 2073-8994
Symmetry 2010, 2, 1745-1762; doi:10.3390/sym2041745 OPEN ACCESS symmetry ISSN 2073-8994 www.mdpi.com/journal/symmetry Review n– Polyanionic Hexagons: X6 (X = Si, Ge) Masae Takahashi Institute for Materials Research, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan; E-Mail: [email protected]; Tel.: +81-22-795-4271; Fax: +81-22-795-7810 Received: 16 August 2010; in revised form: 7 September 2010 / Accepted: 27 September 2010 / Published: 30 September 2010 Abstract: The paper reviews the polyanionic hexagons of silicon and germanium, focusing on aromaticity. The chair-like structures of hexasila- and hexagermabenzene are similar to a nonaromatic cyclohexane (CH2)6 and dissimilar to aromatic D6h-symmetric benzene (CH)6, although silicon and germanium are in the same group of the periodic table as carbon. Recently, six-membered silicon and germanium rings with extra electrons instead of conventional substituents, such as alkyl, aryl, etc., were calculated by us to have D6h symmetry and to be aromatic. We summarize here our main findings and the background needed to reach them, and propose a synthetically accessible molecule. Keywords: aromaticity; density-functional-theory calculations; Wade’s rule; Zintl phases; silicon clusters; germanium clusters 1. Introduction This review treats the subject of hexagonal silicon and germanium clusters, paying attention to the two-dimensional aromaticity. The concept of aromaticity is one of the most fascinating problems in chemistry [1,2]. Benzene is the archetypical aromatic hexagon of carbon: it is a planar, cyclic, fully conjugated system that possesses delocalized electrons. The two-dimensional aromaticity of planar monocyclic systems is governed by Hückel’s 4N + 2 rule, where N is the number of electrons. -

Metallacycle–Based Nickel–Catalyzed Reductive Couplings and Reductive Cross–Electrophile Couplings
Metallacycle–Based Nickel–Catalyzed Reductive Couplings and Reductive Cross–Electrophile Couplings by Amie Renee Frank A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Chemistry) in the University of Michigan 2020 Doctoral Committee: Professor John Montgomery, Chair Associate Professor Amanda L. Garner Associate Professor Corinna S. Schindler Professor John P. Wolfe Amie Renee Frank [email protected] ORCID iD: 0000-0002-6155-3632 © Amie Renee Frank 2020 Dedication To my husband, Caleb Frank. Your love, patience, support and encouragement throughout this adventure makes me want to grow old with you even more. On to the next adventure. ii Acknowledgements I Would like to thank my advisor, Professor John Montgomery, for allowing me the opportunity to Work in his lab the past five years. His guidance and mentorship provided me With the tools necessary to grow as a scientist and develop as a person. Thank you for your support and the freedom to explore my own interests. I Would also like to thank Professor John Wolfe for allowing me the opportunity to rotate in his lab as a first–year graduate student. His support, advice and feedback throughout my graduate career Were alWays greatly appreciated. To the other members on my committee, Professor Corinna Schindler and Amanda Garner, thank you for being inspiring female role models in science. Their support, advice and feedback Were also extremely helpful throughout my graduate career. Additionally, I Would like to thank Professor Anne McNeil for her support as my original research proposal mentor. Lastly, I Would like to thank Professor Brian Coppola and Professor Adam Matzger for their support and advice during times of adversity. -

View of Bicyclic Cobalt Cation 42
University of Alberta Cobalt(III)-Mediated Cycloalkenyl-Alkyne Cycloaddition and Cycloexpansion Reactions by Bryan Chi Kit Chan A thesis submitted to the Faculty of Graduate Studies and Research in partial fulfillment of the requirements for the degree of Doctor of Philosophy Chemistry ©Bryan Chi Kit Chan Spring 2010 Edmonton, Alberta Permission is hereby granted to the University of Alberta Libraries to reproduce single copies of this thesis and to lend or sell such copies for private, scholarly or scientific research purposes only. Where the thesis is converted to, or otherwise made available in digital form, the University of Alberta will advise potential users of the thesis of these terms. The author reserves all other publication and other rights in association with the copyright in the thesis and, except as herein before provided, neither the thesis nor any substantial portion thereof may be printed or otherwise reproduced in any material form whatsoever without the author's prior written permission. Examining Committee Jeffrey M. Stryker, Chemistry, University of Alberta Martin Cowie, Chemistry, University of Alberta Jillian M. Buriak, Chemistry, University of Alberta Frederick G. West, Chemistry, University of Alberta William C. McCaffrey, Chemical Engineering, University of Alberta Lisa Rosenberg, Chemistry, University of Victoria Abstract A comprehensive investigation of cycloalkenyl-alkyne coupling reactions mediated by cobalt(III) templates is presented. The in situ derived cationic η3- cyclohexenyl complexes of cobalt(III) react with some terminal alkynes to afford either η1,η4-bicyclo[4.3.1]decadienyl or η2,η3-vinylcyclohexenyl products, depending on the type and concentration of the alkyne. The mechanism for this cyclohexenyl-alkyne cycloaddition reaction is consistent with the previously reported cobalt-mediated [3 + 2 + 2] allyl-alkyne coupling reaction. -

1. for All Complexes Listed Below, Determine A) Metal Oxidation State B
CH 611 Advanced Inorganic Chemistry – Synthesis and Analysis Practice problems 1. For all complexes listed below, determine a) metal oxidation state b) total number of electrons contributed from metal c) total number of electrons contributed from the ligand set d) total electron count of the complex Please note: use the ionic model unless asked otherwise and comment on any complexes that do not obey the 18VE rule or have CN < 6. 5 i) ( ‐Cp)2Fe 5 ii) [( ‐Cp)2Co] Ionic Model Metal oxidation state: 3+ 3+ Co Metal electron count: 6 Ligand electron count: 6 + 6 Total electron count: 18 iii) Co2(CO)8 CH 611 Advanced Inorganic Chemistry – Synthesis and Analysis Practice problems 2 iv) Ru( ‐en)2H2 v) Mn(4‐salen)Cl Cl Ionic Model N N Metal oxidation state: 3+ Mn3+ Metal electron count: 4 O O Ligand electron count (s only):2+2+2+2+2 Total electron count: 14 A total electron count at the metal of just 14 electrons is predicted using the ionic model but only considering ‐bonds. Both oxide and chloride ligands are capable of ‐donation to the empty metal orbitals making up the 4 extra electrons to comply with the 18VE rule. The nature of this ‐donation can be deciphered using the molecules point group symmetry, the corresponding character table and the method of systematic reduction of non‐shifted/inverted ‐vectors. vi) Ti(iso‐propoxide)4 A total electron count at the metal of just 8 electrons is predicted using the ionic model but only considering ‐bonds. Each isopropoxide ligand is capable of ‐donation to the empty metal orbitals making up the 10 extra electrons to comply with the 18VE rule.