The University of Chicago Integrative Genetic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Girl Names Registered in 1996
Baby Girl Names Registered in 1996 # Baby Girl Names # Baby Girl Names # Baby Girl Names 1 Aaliyah 1 Aiesha 1 Aleeta 1 Aamino 2 Aileen 1 Aleigha 1 Aamna 1 Ailish 2 Aleksandra 1 Aanchal 1 Ailsa 3 Alena 2 Aaryn 4 Aimee 1 Alesha 1 Aashna 1Ainslay 1 Alesia 5 Abbey 1Ainsleigh 1 Alesian 1 Abbi 4Ainsley 6 Alessandra 3 Abbie 1 Airianna 1 Alessia 2 Abbigail 1Airyn 1 Aleta 19 Abby 4 Aisha 5 Alex 1 Abear 1 Aishling 25 Alexa 1 Abena 6 Aislinn 1 Alexander 1 Abigael 1 Aiyana-Marie 128 Alexandra 32 Abigail 2Aja 2 Alexandrea 5 Abigayle 1 Ajdina 29 Alexandria 2 Abir 1 Ajsha 5 Alexia 1 Abrianna 1 Akasha 49 Alexis 1 Abrinna 1Akayla 1 Alexsandra 1 Abyen 2Akaysha 1 Alexus 1 Abygail 1Akelyn 2 Ali 2 Acacia 1 Akosua 7 Alia 1 Accacca 1 Aksana 1 Aliah 1 Ada 1 Akshpreet 1 Alice 1 Adalaine 1 Alabama 38 Alicia 1 Adan 2 Alaina 1 Alicja 1 Adanna 1 Alainah 1 Alicyn 1 Adara 20 Alana 4 Alida 1 Adarah 1 Alanah 2 Aliesha 1 Addisyn 1 Alanda 1 Alifa 1 Adele 1 Alandra 2 Alina 2 Adelle 12 Alanna 1 Aline 1 Adetola 6 Alannah 1 Alinna 1 Adrey 2 Alannis 4 Alisa 1 Adria 1Alara 1 Alisan 9 Adriana 1 Alasha 1 Alisar 6 Adrianna 2 Alaura 23 Alisha 1 Adrianne 1 Alaxandria 2 Alishia 1 Adrien 1 Alayna 1 Alisia 9 Adrienne 1 Alaynna 23 Alison 1 Aerial 1 Alayssia 9 Alissa 1 Aeriel 1 Alberta 1 Alissah 1 Afrika 1 Albertina 1 Alita 4 Aganetha 1 Alea 3 Alix 4 Agatha 2 Aleah 1 Alixandra 2 Agnes 4 Aleasha 4 Aliya 1 Ahmarie 1 Aleashea 1 Aliza 1 Ahnika 7Alecia 1 Allana 2 Aidan 2 Aleena 1 Allannha 1 Aiden 1 Aleeshya 1 Alleah Baby Girl Names Registered in 1996 Page 2 of 28 January, 2006 # Baby Girl Names -

Rates and Patterns of Indels in HIV-1 Gp120 Within and Among Hosts
Western University Scholarship@Western Electronic Thesis and Dissertation Repository 8-19-2020 1:00 PM Rates and patterns of indels in HIV-1 gp120 within and among hosts John Lawrence Palmer, The University of Western Ontario Supervisor: Poon, Art F.Y., The University of Western Ontario A thesis submitted in partial fulfillment of the equirr ements for the Master of Science degree in Pathology and Laboratory Medicine © John Lawrence Palmer 2020 Follow this and additional works at: https://ir.lib.uwo.ca/etd Part of the Molecular Genetics Commons Recommended Citation Palmer, John Lawrence, "Rates and patterns of indels in HIV-1 gp120 within and among hosts" (2020). Electronic Thesis and Dissertation Repository. 7308. https://ir.lib.uwo.ca/etd/7308 This Dissertation/Thesis is brought to you for free and open access by Scholarship@Western. It has been accepted for inclusion in Electronic Thesis and Dissertation Repository by an authorized administrator of Scholarship@Western. For more information, please contact [email protected]. Abstract Insertions and deletions (indels) in the HIV-1 gp120 variable loops modulate sensitivity to neutralizing antibodies and are therefore implicated in HIV-1 immune escape. However, the rates and characteristics of variable loop indels have not been investigated within hosts. Here, I report a within-host phylogenetic analysis of gp120 variable loop indels, with mentions to my preceding study on these indels among hosts. We processed longitudinally-sampled gp120 sequences collected from a public database (n = 11,265) and the Novitsky Lab (n=2,541). I generated time-scaled within-host phylogenies using BEAST, extracted indels by reconstructing ancestral sequences in Historian, and esti- mated variable loop indel rates by applying a Poisson-based model to indel counts and time data. -

For a Casual Faith and This Is No Time to Go It Alone
NO TIME UNITARIAN UNIVERSALIST ASSOCIATION Annual Report FOR A Fiscal Year 2018 CASUAL FAITH TABLE OF CON- TENTS A letter from Rev. Susan Frederick-Gray 1 Time to... Equip Congregations for Health and Vitality 4 Train and Support Leaders 10 Advance UU Values and Justice 14 Organizational and Institutional Change 18 Grow New Congregations and Communities 22 Leadership 23 Financial Performance 24 Contributors 26 Congregations Individuals Legacy Society In memorium 76 Beacon Press and Skinner House 79 Our Unitarian Universalist Principles 80 Two themes came to define my first year as your UUA President – This is TABLE No Time for a Casual Faith and This is No Time to go it Alone. This is a defining time in our nation and for our planet. The challenges, opportunities and crises that mark this time impact our own lives and our congregations and communities. Unfortunately, in times of crises and change None of this could happen without your OF CON- — when rhetoric of fear and defensiveness collective support, as congregations and dominate — it is all too common for people individuals. The UUA is the embodiment and institutions to break down, or to turn of the covenant we make to each other as inward and protective. But it is precisely in Unitarian Universalists to build something times of change and urgency when we need stronger than any of us could be alone. more courage, more love, more commitment When the UUA shows up for congregations in order to nurture the hope that is found following hurricanes and wildfires, when in seeing the possibilities that live within we help congregations find and call new TENTS humanity and community. -

Dean's List| Spring 2020
DEAN’S LIST| SPRING 2020 Dean's List requirements: Students must complete 12 or more credits with a semester GPA of 3.6-4.0 and have a cumulative GPA of at least 2.0. This printed list does not include students who have requested their records remain confidential. Aagard, Makennah C. Adams, Samuel R. Alexander, Alyssa M. Aagard, Nathan Adams, Siera D. Alexander, Braden Aamodt, Alexis K. Adams, Torri R. Alexander, Stephen J. Abad, Gabriela Adamson Mock, Jennifer L. Alexander, Tatum Abbott, Madison N. Adamson, Calli L. Aley, Camille S. Abbott, Michael Adamson, Carlee Alfonso, Buen Rafael Abbott, Shelby R. Adamson, Kailee A. Alfonso, Jasmine A. Abbott, Tyler Addison, Emily A. Alford, Daniel E. Abdrisaeva, Meerim Adkins, Chandler M. Alger, Jeremy H. Ableman, Darby K. Adkins, Melissa M. Alghanmi, Abdulrahman Abplanalp, Tanden R. Adkins, Skyler J. Alghanmi, Byan A. Abrahamson, Samantha Affleck, Eirik Ali, Mohamed R. Abrams, Kayla M. Afinogenova, Kateryna Alkathir, Majed W. Acerson, Stephanie J. Aguayo, Giselle N. Allan, Abigail Acker, Steven J. Aguayo, Heidi Allan, Kaelan E. Ackerman, Lauren A. Aguero, Christine C. Allan, Nicholas B. Ackerson, Julie A. Aguilar-Bojorquez, Margarita Allan, Wyatt H. Ackley, Chad Aguirre Corral, Marlenne Allen, Angela I. Acosta, Alexis Ah Mu, Tessa Allen, Ashleigh J. Acosta, Christian J. Ahlborn, Katelynn T. Allen, Audrey N. Acosta, Gerardo M. Ahlman, Susanne A. Allen, Brittnie Acosta, Kevin Ahlstrom, Hannah Allen, Christina E. Adair, Danielle M. Aiello, Austin S. Allen, Claire E. Adair, Robert B. Aikin-Arendt, Alex Allen, Colby K. Adamic, Jonathan D. Akana, Ethan S. Allen, Erin M. Adams, Abigail M. Akers, Nathanial S. -

2020 Unclaimed Property Master List for WEB PUBLISHING.Xlsx
TARRANT COUNTY UNCLAIMED PROPERTY As of November 2020 Name 7-11 1 FIRST UNITED 1015 RANDOLL MILL ASSOC INC 114 & MAIN PARTNERS LP 2013B PROPERTY OWNER LLC & SILVER BAY OPERATING 34 CROSS LLC 4L PLUMBING INC A A A B S INC A B C GROUP INC A C PROPERTIES A L & VELINDA CLEVINGER A R A HOLDING OR NEITA CAMPBELL A TO Z OFFICE SUPPLY A-AAA MOBILE KEY SERVICE AAYA INC ABBOTT, MARK WAYDE ABDULLAH, TYMR I ABDULMAKSOUD, EHAB ABDULMUJEEB, NADIA ABELL, NAN ELIZABETH ABODE TITLE ABOUELNAGA, ASHRAF ABRAMS & ABRAMS ABRAMS, ATHLENE ACCENT AWARDS INC ACCENTS & GIFTS ACE CASTERS, INC ACE HARDWARE ACEVEDO, MARIA TERESA ACOFF, AMOS ACON, TALENA ACOSTA, ERICA ETVIR PAUL ACOSTA, PAULA R ACQUISITIONS CONSULTING CORP ACTION RENT TO OWN ACTION RENT TO OWN ACTION TV ACTION VACUUM ADAM M BREST, ADAM M ADAM, JOHN F JR ETUX MARY H ADAME, JESUS ETUX ARTEMIA ADAME, TERESITA & ROSA ADAMS, BELINDA L ADAMS, PAMELA A ADAMS, ALICIA J ADAMS, ANTHONY T ADAMS, BRIAN TARRANT COUNTY UNCLAIMED PROPERTY As of November 2020 Name ADAMS, FRANK ADAMS, HENRY C ADAMS, JAMES E ADAMS, TIMOTHY ADAMS, VESTA M ADDISON, BERNICE ADEDIRE, MODINAT ADEL, RIZK ABDELMASEEH ADKINS & MEYERS ADKINS, ALLEN L ADKINS, DEBORAH ANN ADKINSON, ARLISA ADLER SEWING CENTERS ADMINISTRATOR VETERAN AFFAIRS ADMIRE, VICKIE J ADOBE TITLE, LLC ADORNO YOSS WHITE & WIGGINS ADRIAN, JAMES PHILLIP ADRIANE WHITLEY & W T TRANSPORT ADTANI, MINAZ ADVANCED APARTMETN MOVERS ADVANCED IT SOLUTIONS ADVANCED MEDICAL PARTNERS INC ADVANTAGE BUYERS CLUB AEROEJECTIVO, A. A., DBA AEROE AFANDE, FARDOUS AFFILIATED BANK AFFILIATED COMPUTER SERVICES INC AGEE, RICKY AGEMBO, TOM O. -

Lexington Countg, SC 1
FEMALE INDEX TO MARRIAGES - Lexington Countg, S. C. PAGE Eniy COn PHONE BOOK — DIRECTORY MADE IY THE OHT INPEX COMPANY, COLUMOI, OHIO ( 001 001 001 e I!M H*. y COn DATA PROCESSED INDEXES 1 LICENSE DATE ISSUED DATE OF MARRIAGE FEMALES MALES PERSON OFFICIATING NUMBER SURNAME GIVEN NAME AGE RACE Ho. Day Year Mo- Day Year 82-1060 AARON CONNIE STEWART 41 AARON JAMES GORDON W 11 11 1982 11 19 1982 REV WAYNE YOUNG LEXINGTON SC LEXINGTON SC LEXINGTON SC 82-1112 AARON CONNIE STEWART 41 AARON JAMES GORDON w 12 / 1982 12 8 1982 JOHN LAND JOHNSON PROS JD LEXINGTON SC LEXINGTON SC LEXINGTON SC 86-0084 AARON MARY JO 39 BUSH ROY EUGENE W 2 4 1986 2 9 1986 REV JOHN 'L SATTERWHITE WEST COLUMBIA SC LEXINGTON SC WEST COLUMBIA SC - ******* 85-0247 A8BETT LENA FAYE 41 ELVINGTON RICHARD LYNDELLE W 4 8 1985 4 12 1985 DAVID F WALDRQP SR NP LEXINGTON SC LEXINGTON SC LEXINGTON COUNTY SC J ******* 85-^0935 ABBOTT BETTY ANN 40 STRANGE FRAIiKIE BENJAMIN W 10 2 1985 10 4 1985 REV JOHN-' H MCKEOWN CAYCE SC CAYCE SC WEST COLUMBIA SC j 82-0625 ABBOTT DEMISE MAE 24 StALY VIRGIL flUINN W 7 6 1982 7 31 1982 REV C P FISHER II WEST COLUMBIA COLUMBIA SC LEXINGTON COUNTY SC 81*1068 ABBOTT JUDITH LYNN 24 STURKIE DONALD UYMAN W 11 24 1981 12 22 1981 JOHN LAND JOHNSON PRO JUDGE WEST COLUMBIA SC WAGENER SC LEXINGTON SC 82-0337 ABBOTT RAt CHRISTINE 29 ABBOTT BRUCE * W 4 16 1982 4 23 1982 J R SWYGERT ASC PRO JOG CAYCE SC CAYCE SC LEXINGTON SC _ ******* 84-1043 ACKERMAN MARGARET P 45 HENDRIX SR ROY PAUL W 10 23 1984 1 11 1985 JOHN LAND JOHNSON PRO JD WEST COLUMBIA SC GILBERT SC LEXINGTON -

Bettendorf High School 1St Semester Honor Roll 2014-15 Grade 11
Bettendorf High School 1st Semester Honor Roll 2014-15 Grade 11 Highest Honor Roll – 4.00 Anna Baker Haley Humphrey Karlee Scheib Isabella Blackman Alexa Ito Lindsey Sheldon Katherine Braught Amber Jerson Aaron Silva Andrew Doyle Makayla Kuhrt Caroline Stevens Kennedy Gagne Ian Levetzow Scott Summers Brandon Gore Alyssa Lira Isaac Thompson Andrea Hale Amanda Matthews Michael Trahan Alyssa Henson Amanda Myers Shanda Westerkamp Luke Hovey Grant Nickles High Honor Roll - 3.999 – 3.500 Madison Albracht Samuel Hall Joshua Pauley Virginia Allen Emily Hatch Bailey Pribyl Logan Anderson Max Hollis Sarah Rasing John Bai Gabriella Houk Taylor Regenwether Ian Beck Micah Jacks Owen Ridenour Claire Bryant Paige Kelly Macy Rinehart Courtney Bush Charlotte Koberg Lauren Ross Michael Carr Sarah Lammers Margaret Runge Noah Clark Ellie Lampe Elizabeth Saathoff Samantha Connor Zachary Lane Alexandra Salter Carter Cook Jordan Laufenberg Alex Schmertmann Brandon Cooley Justin Lemasters Megan Sharkey Mitchell Darrow Karly Lent Sydney Singh Erica Eastland Alexander Locey Emilia Skeen Julia Elias Blaire Logan David Smidt Max Erpelding Kendra Marr Matthew Smith Elliott Evans Joshua McDowall Tyler Smith Connor Folkman Dalton McLaughlin Shana Snyder Dennis Fredell Lucas Miller Kaley Sortillo Robert Gallagher Carly Mitchell Anastasia Sperling McKenzie Gamble Emily Morris Hannah Sunderman Brenna Hafner Charlyn Moss Sergei Swart Michelle Hafner Olivia Osborn Ashley Swearengen Isabelle VanLandschoot John Wells Jadwin Woodall Marika Vi Kayli Whitcanack Nolan Wright -

Directory of Teacher Email Addresses
Building Name E_MAIL Last Name First Name Position Name AEROSPACE AND NAT SCIENCE ACAD [email protected] AHIEKPOR KRISTIN INTERVENTION SPECIALIST AEROSPACE AND NAT SCIENCE ACAD [email protected] COUSINO JENNIFER ENGLISH HS AEROSPACE AND NAT SCIENCE ACAD [email protected] DAUNHAUER AMY SENIOR DIRECTOR AEROSPACE AND NAT SCIENCE ACAD [email protected] DEBERG-PEACE MONICA ASST DIRECTOR ANSAT AEROSPACE AND NAT SCIENCE ACAD [email protected] ELLIS KENDRA ENGLISH HS AEROSPACE AND NAT SCIENCE ACAD [email protected] HOLMAN WILLIAM T AND I AEROSPACE AND NAT SCIENCE ACAD [email protected] HOOKER MELISSA SCIENCE HS AEROSPACE AND NAT SCIENCE ACAD [email protected] JURKIEWICZ CASSANDRA SEC IV 10.5 M 8 HR AEROSPACE AND NAT SCIENCE ACAD [email protected] KINNER ANTHONY SOCIAL STUDIES HS AEROSPACE AND NAT SCIENCE ACAD [email protected] NABER-FISHER GLENELLYN FOREIGN LANGUAGE AEROSPACE AND NAT SCIENCE ACAD [email protected] NEUHAUS TIMOTHY PHYS ED HS AEROSPACE AND NAT SCIENCE ACAD [email protected] SANZENBACHER LINDSAY SCIENCE HS AEROSPACE AND NAT SCIENCE ACAD [email protected] SMITH ROBERT INTERVENTION SPECIALIST AEROSPACE AND NAT SCIENCE ACAD [email protected] SWAN CHRISTINE SOCIAL STUDIES HS AEROSPACE AND NAT SCIENCE ACAD [email protected] WALLER AUSTIN MATH HS AEROSPACE AND NAT SCIENCE ACAD [email protected] YOUNG MAURICE MATH HS ARLINGTON ES [email protected] ACOSTA WENDI KINDERGARTEN ARLINGTON ES [email protected] BARNES KORA CCSE ARLINGTON ES [email protected] BERMEJO CARMEN ELEMENTARY ADMIN ASSIST ARLINGTON ES [email protected] BLUST NATHAN ASST PRINCIPAL ARLINGTON ES [email protected] -
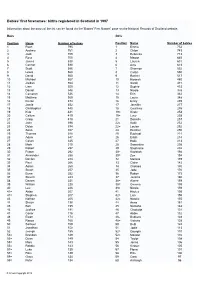
Babies' First Forenames: Births Registered in Scotland in 1997
Babies' first forenames: births registered in Scotland in 1997 Information about the basis of the list can be found via the 'Babies' First Names' page on the National Records of Scotland website. Boys Girls Position Name Number of babies Position Name Number of babies 1 Ryan 795 1 Emma 752 2 Andrew 761 2 Chloe 743 3 Jack 759 3 Rebecca 713 4 Ross 700 4 Megan 645 5 James 638 5 Lauren 631 6 Connor 590 6 Amy 623 7 Scott 586 7 Shannon 552 8 Lewis 568 8 Caitlin 550 9 David 560 9 Rachel 517 10 Michael 557 10 Hannah 480 11 Jordan 554 11 Sarah 471 12 Liam 550 12 Sophie 433 13 Daniel 546 13 Nicole 378 14 Cameron 526 14 Erin 362 15 Matthew 509 15 Laura 348 16 Kieran 474 16 Emily 289 17 Jamie 452 17 Jennifer 277 18 Christopher 440 18 Courtney 276 19 Kyle 421 19= Kirsty 258 20 Callum 419 19= Lucy 258 21 Craig 418 21 Danielle 257 22 John 396 22= Katie 252 23 Dylan 394 22= Louise 252 24 Sean 367 24 Heather 250 25 Thomas 348 25 Rachael 221 26 Adam 347 26 Eilidh 214 27 Calum 335 27 Holly 213 28 Mark 310 28 Samantha 208 29 Robert 297 29 Stephanie 202 30 Fraser 292 30= Kayleigh 194 31 Alexander 288 30= Zoe 194 32 Declan 284 32 Melissa 189 33 Paul 266 33 Claire 182 34 Aaron 260 34 Chelsea 180 35 Stuart 257 35 Jade 176 36 Euan 252 36 Robyn 173 37 Steven 243 37 Jessica 160 38 Darren 231 38= Aimee 159 39 William 228 38= Gemma 159 40 Lee 226 38= Nicola 159 41= Aidan 207 41 Hayley 156 41= Stephen 207 42= Lisa 155 43 Nathan 205 42= Natalie 155 44 Shaun 198 44 Anna 151 45 Ben 195 45 Natasha 148 46 Joshua 191 46 Charlotte 134 47 Conor 176 47 Abbie 132 48 Ewan 174 -

Name City State Lilian Achayo Portland ME Charlyn Achola Wells ME Karli Rae Adams Parsonsfield ME Rachida A
SMCC Fall 2018 Dean's List Name City State Lilian Achayo Portland ME Charlyn Achola Wells ME Karli Rae Adams Parsonsfield ME Rachida A. Adour Portland ME Justin Michael Ahearn Lebanon ME Erik Dean Aikins Portland ME Jennifer Lee Ainsworth Lebanon ME Diandra Audrey Ajemian Portland ME Banah Labed Al Hanfy Portland ME Joel Alcide Westbrook ME Ali H. Alhameedi South Portland ME Labed Jalal Alhanfy South Portland ME Jumana Labed Al-Hanfy South Portland ME Ian Michael Allard Scarborough ME Joshua Coleman Allen Falmouth ME Melinda Louise Allen Portland ME Isak Andre Almeida Silva Old Orchard Beach ME Greta Rae Alsfeld N Waterboro ME Martin Daniel Altieri South Portland ME Hernan S. Alvarez Portland ME Adam Joseph Amabile Portland ME Daniele Amandolini Portland ME Chael Benedicto Anastacio Buxton ME Victoria Lee Anderson Gray ME Jamie Alyse Anfinson Falmouth ME Paul Joseph Apperson Westbrook ME David Ernest Applin Berwick ME Madison Lane Applin North Berwick ME Bryn Arbuckle Portland ME Leslie Naty Arias Duran Somersworth NH Alexandra Victoria Armstrong Portland ME Sean Francis Arsenault Scarborough ME Corey Thomas Ash York ME Charles Athanasion Portland ME Michele Renee Atkins North Yarmouth ME Katherine Joan Atwood Cumberland ME Elijah Gabriel Austin Pownal ME Nathan Alexander Austin Harpswell ME Lena Azama Portland ME Dawn Rebecca Babb Limerick ME Tara Nicole Balch Portland ME Anthony James Balko Portland ME Kevin Thomas Ball Jaffrey NH Brian Anthony Ballzigler Scarborough ME Jordan Lorraine Bantz Oxford ME Katrina L. Barbaria Saco ME Benjamin Joshua Bard Sidney ME Jean Pierre Rwaka Barengayabo Biddeford ME Cyrus N. Barker Portland ME Ashlynn Glenna Barnes South Portland ME Zachery Tyler Barrows Standish ME Grace E. -

MINUTES of the MEETING of the BOARD of REGENTS of the UNIVERSITY SYSTEM of GEORGIA HELD at Clayton College & State University in Morrow, Georgia October 7 and 8, 1997
MINUTES OF THE MEETING OF THE BOARD OF REGENTS OF THE UNIVERSITY SYSTEM OF GEORGIA HELD AT Clayton College & State University in Morrow, Georgia October 7 and 8, 1997 CALL TO ORDER The Board of Regents of the University System of Georgia met on Tuesday, October 7 and Wednesday, October 8, 1997 in room 203, the Meeting Room, of the Harry S. Downs Continuing Education Center at Clayton College & State University in Morrow, Georgia. The following Committees of the Board of Regents met in succession on Tuesday, October 7: the Audit Committee; the Teaching Hospital Committee; the Committee on Finance and Business Operations; the Committee on Real Estate and Facilities; the Committee on Education, Research, and Extension; and the Committee on Organization and Law. The Chair of the Board, Regent S. William Clark, Jr., called the meeting to order at 9:00 a.m. on Wednesday, October 8. Present on Wednesday, in addition to Chair Clark, were Vice Chair Edgar L. Jenkins and Regents Thomas F. Allgood, Sr., Shannon L. Amos, David H. (Hal) Averitt, Juanita P. Baranco, Kenneth W. Cannestra, A. W. “Bill” Dahlberg, George M. D. (John) Hunt III, Charles H. Jones, Donald M. Leebern, Jr., Elridge W. McMillan, Edgar L. Rhodes, and Glenn S. White. INVOCATION The invocation was given on Wednesday, October 8 by Mr. Zachary Ross, President of the Student Government Association at Clayton College & State University. ATTENDANCE REPORT The attendance report was read on Wednesday, October 8 by Secretary Gail S. Weber, who announced that Regent A. W. “Bill” Dahlberg had asked for and been given permission to be absent on Tuesday, October 7, 1997 and that Regents J. -
![NLN Mouse Monoclonal Antibody [Clone ID: OTI1D6] Product Data](https://docslib.b-cdn.net/cover/9372/nln-mouse-monoclonal-antibody-clone-id-oti1d6-product-data-2099372.webp)
NLN Mouse Monoclonal Antibody [Clone ID: OTI1D6] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for CF504178 NLN Mouse Monoclonal Antibody [Clone ID: OTI1D6] Product data: Product Type: Primary Antibodies Clone Name: OTI1D6 Applications: FC, IF, IHC, WB Recommended Dilution: WB 1:500~2000, IHC 1:150, IF 1:100, FLOW 1:100 Reactivity: Human, Monkey, Mouse, Rat Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Full length human recombinant protein of human NLN(NP_065777) produced in HEK293T cell. Formulation: Lyophilized powder (original buffer 1X PBS, pH 7.3, 8% trehalose) Reconstitution Method: For reconstitution, we recommend adding 100uL distilled water to a final antibody concentration of about 1 mg/mL. To use this carrier-free antibody for conjugation experiment, we strongly recommend performing another round of desalting process. (OriGene recommends Zeba Spin Desalting Columns, 7KMWCO from Thermo Scientific) Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 80.5 kDa Gene Name: Homo sapiens neurolysin (NLN), mRNA; nuclear gene for mitochondrial product. Database Link: NP_065777 Entrez Gene 75805 MouseEntrez Gene 117041 RatEntrez Gene 696884 MonkeyEntrez Gene 57486 Human Q9BYT8 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 4 NLN Mouse Monoclonal Antibody [Clone ID: OTI1D6] – CF504178 Background: This gene encodes a member of the metallopeptidase M3 protein family that cleaves neurotensin at the Pro10-Tyr11 bond, leading to the formation of neurotensin(1-10) and neurotensin(11-13).