High-Throughput SNP Discovery and Genotyping for Constructing a Saturated Linkage Map of Chickpea (Cicer Arietinum L.)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ICAB Monthly News Bulletin June 2021 Issue
June 2021 Number 377 icab news bulletin Monthly News Briefing from the Institute of Chartered Accountants of Bangladesh (ICAB) SME’s Contribution to GDP is Very Low in Comparison to Other Emerging Economies, Needs SMPs’ Support to Revive the Sector Speakers suggest at a Global Webinar SME’s Contribution to 1 GDP... Revive the Sector Auditing Software 3 Launched:.. audit begins President’s 4 Communication Inauguration of ICAB 7 Digital... Education Minister Some provisions of 8 Companies... Webinar Meeting with NBR 9 Chairman Meeting with CAG 9 Bangladesh Press Conference on 10 National Budget 2021 ICAB Team paid a 12 courtesy... Secretary, GoB Campus News 12 Chartered Accountancy- 13 A Career Choice Training on “Minimum 13 audit... each audit client” Virtual Members’ 14 Conference... forward’ Online Certificate Course 14 ... Audit (IS Audit)-5th Batch n Bangladesh SMEs are considered as situation has aggravated as cash-flow problem, Online Certificate Course 14 on... Based on ISA engine of economic growth that constitutes liquidity crisis, IT skill gap, reduction of Anwaruddin Chowdhury 15 I over 90 percent of business enterprises while production costs, shifting to e-commerce Participates... Meeting it is 97.60 percent in India, 99 percent in China, practices and digitalization of operations have Admission as Fellows 15 99.70 percent in Japan and 60 percent in emerged as new challenges to SME sector. Permission to Continue 15 Pakistan. SME contributes to GDP 20.25 Practice The Institute of Chartered Accountants of percent which is very low in comparison to other Permission to Start 16 Bangladesh (ICAB) organized the Global emerging economies. -

Annual Report Have Been Captured in the Campus of NIAB Annual Report | 2019-20
Annual NIAB Report 2019-20 Photographs used in the Annual Report have been captured in the campus of NIAB Annual Report | 2019-20 1 2 Annual Report | 2019-20 S. No. TABLE OF CONTENTS Page No. 1. Mission and Vision of NIAB 05 2. From the desk of Director 06 3. Research Projects: 09 A. Animal Genomics and Reproduction 09 a. Livestock Genomics for Cattle Improvement and Transgenic Farmed Animals (Dr. 10 Subeer S Majumdar) b. Reproductive Biology, Gametogenesis, Oocyte atresia, DNA damage response and 15 repair pathways (Dr. H.B.D. Prasada Rao) c. Biopharming Using Farmed Animals and Avenues for Obtaining Sperm with Elite 19 Trait (Dr. Nirmalya Ganguli) d. Aptamer and antibody based point-of-care diagnostics for better animal production 22 and health ( Dr. Pankaj Suman) B. Animal Health 26 a. Microbial Patho biology and One Health (Dr. Nagendra R. Hegde) 27 b. Understanding the virulence mechanisms of the zoonotic pathogen, Brucella and 33 development of improved vaccines and diagnostic assays for animal and human brucellosis (Dr. Girish K Radhakrishnan) c. Development of Leptospirosis vaccine and novel veterinary adjuvants (Dr. Syed M 36 Faisal) d. Host Pathogen Interaction Studies on Animal and Avian Viruses (Dr. Madhuri 39 Subbiah) e. Host pathogen interaction studies on animal parasites (Dr. Anand Srivastava) 42 f. Study of Virulence, Antimicrobial Resistance and Host Pathogenesis during 47 Intracellular Pathogens infection (Dr. Paresh Sharma) g. Role of CDK-related kinases (Crks) in transcription regulation in Toxoplasma gondii 51 (Dr. Abhijit S Deshmukh) h. Tuberculosis and other zoonotic diseases of livestock: Molecular pathogenesis and 55 Intervention Strategies (Dr. -
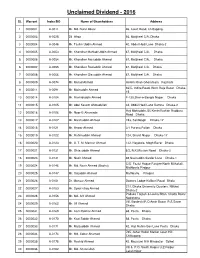
Unclaimed Divident
Unclaimed Dividend - 2016 SL Warrant Index/BO Name of Shareholders Address 1 0000001 A-0011 Mr. Md. Nurul Absar 46, Court Road, Chittagong. 2 0000003 A-0025 Dil Afroz 66, Motijheel C/A, Dhaka 3 0000004 A-0048 Mr. Taslim Uddin Ahmed 40, Abdul Hadi Lane Dhaka-2 4 0000005 A-0053 Mr. Khondker Mahtab Uddin Ahmed 67, Motijheel C/A. Dhaka 5 0000006 A-0054 Mr. Khondker Raisuddin Ahmed 67, Motijheel C/A. Dhaka 6 0000007 A-0055 Mr. Khondker Raziuddin Ahmed 67, Motijheel C/A. Dhaka 7 0000008 A-0056 Mr. Khondker Giasuddin Ahmed 67, Motijheel C/A. Dhaka 8 0000009 A-0074 Mr. Murad Ahmed Hetem Khan Ghoramara Rajshahi 66/C, Indira Road, West Raja Bazar Dhaka- 9 0000011 A-0091 Mr. Moinuddin Ahmed 15 10 0000014 A-0104 Mr. Rashiduddin Ahmed F-130,Sher-e-Bangla Nagar Dhaka 11 0000015 A-0105 Mr. Abul Kasem Ahmadullah 34, Abdul Hadi Lane Ramna Dhaka-2 Haji Mohiuddin, 56 KiminiVushan Rudduru 12 0000016 A-0106 Mr. Noor-E-Ahameda Road Dhaka 13 0000017 A-0107 Mr. Masihuddin Ahmed 193, Santibagh Dhaka-17 14 0000018 A-0121 Mr. Anwar Ahmed 3/1 Purana Paltan Dhaka 15 0000019 A-0122 Mr. Rahimuddin Ahmed 124, Shanti Nagar Dhaka-17 16 0000020 A-0123 Mr. A. T. M. Mansur Ahmed 132, Nayatola, Mogh Bazar Dhaka 17 0000021 A-0131 Mr. Ghiasuddin Ahmed 8/2, R.K.Mission Road Dhaka-3 18 0000023 A-0141 Mr. Nazir Ahmed 48,Nasiruddin Sardar Lane Dhaka-1 C/O. Fazlul Haque Farajee North Mithakali, 19 0000024 A-0146 Mr. -

ICAB Monthly News Bulletin July 2020 Issue
Number 366 July 2020 ICAB NEWS BULLETIN Monthly News Briefing from the Institute of Chartered Accountants of Bangladesh (ICAB) Strongest and Resilient Economies Need Trustworthy Information Strongest and Resilient 1 Economies... Information CRC Donates Medical 3 Equipment... COVID-19 President’s Communication 4 Trustworthy Financial 6 Reporting... Conference Virtual SAFA Forum on 7 Challenges... Outbreak Recommendations for 7 Monetary Policy Virtual Members’ 8 Conference... Challenges Sabbir Ahmed Participated 9 in ICMA... Webinar he strongest and most resilient Salman F Rahman MP, Private Sector Industry Admission as Fellows 9 economies will be those where people and Investment Adviser to the Honorable Prime T can trust information they receive; the Minister of the Government of the People’s Permission to Start 9 leaders can make well-informed decisions, Republic of Bangladesh attended the Practice businesses can be held to account and public conference as the Chief Guest. Manil Permission to Continue 9 finances are transparent. The skills that the Jaysinghe FCA, President CA Sri Lanka was Practice accountancy profession instills in measuring, present as Special Guest while Dr. In-Ki Joo, reporting and assuring –will be fundamental in President of the International Federation of Permission to Join as 9 Accountants (IFAC) was present as Guest of Partner building a World of Strong Economies. The technical skills, ethical approach and the Honour. Robert Hodgkinson FCA, Executive Permission to Open CA 10 integrity are very important for the purpose. Director, Technical at the Institute of Chartered Firm Under the current pressures of the pandemic, Accountants in England and Wales (ICAEW), Permission to train Articled 10 everybody CAs, in particular should take UK presented the keynote paper. -

2013 ANNUAL REPORT I Am a Little Obsessed with Fertilizer
2013 ANNUAL REPORT I am a little obsessed with fertilizer. I mean I’m fascinated with its role, “not with using it. Two out of every five people on Earth today owe their lives to the higher crop outputs that fertilizer has made possible. It helped fuel the Green Revolution, an explosion of agricultural productivity that lifted hundreds of millions of people around the world out of poverty.” – Bill Gates, “Here’s My Plan to Improve Our World and How You Can Help,” November 12, 2013 CONTENTS 2 IFDC Around the World 4 2014 IFDC Board of Directors Message from the Chairman of the Board and the 5 President and Chief Executive Officer 40 Years: Amidst Evolution, Focus on Smallholder 6 Farmers Remains 10 Highlights 12 Annual Activities Align With Strategic Plan 16 2013 Strategy Workshops 20 2013 Special Initiatives 22 EurAsia Division 26 East and Southern Africa Division 32 North and West Africa Division 38 Office of Programs 42 Training & Workshop Coordination Unit 44 2013 Selected Articles, Presentations and Studies 46 IFDC Offices and Staff 57 Revenue Sources 58 Financial Highlights 60 Acronyms and Abbreviations and Publication Credits ANNUAL REPORT 2013 | 1 Countries With IFDC Projects in 2013 Bangladesh Benin Burkina Faso Burundi Cape Verde Chad Côte d’Ivoire Democratic Republic of Congo Ethiopia Gambia Ghana Guinea Guinea-Bissau Kenya Kyrgyzstan Liberia Mali Mozambique Niger Nigeria Rwanda Senegal Sierra Leone South Sudan Tajikistan Tanzania Togo Uganda United States of America IFDC Zambia Zimbabwe Around the World 2 | 2013 ANNUAL REPORT IFDC Around the World For a complete list of countries in which IFDC has worked, visit www.ifdc.org/About/Map_IFDC_in_the_World. -

Fazlul Huq and Bengal Politics in the Years Before the Second Partition of Bengal (1947)
6 Fazlul Huq and Bengal Politics in the years before the Second Partition of Bengal (1947) On 24 April 1943, H.E. the Governor Sir John Herbert with the concurrence of the Viceroy, issued a proclamation by which he revoked the provision of Section 93 and invited the leader of the Muslim League Assembly Party, Khwaja Sir Nazimuddin to form a new ministry. The aspirant Nazimuddin who made constant effort to collect support in order to form a new ministry, ultimately managed to have the support of 140 members in the Bengal Assembly (consisting of 250 membrs). On the other hand, the Opposition (led by Fazlul Huq) had 108 members. Following the dismissal of the Second Huq Ministry, a large number of Muslim M.L.A.s thought it expedient to cross the floor and join the League because of Governor‟s inclination towards Nazimuddin whom he invited to form a new ministry. As they flocked towards the League, the strength of the League Parliamentary Party rose from 40 to 79. Abul Mansur Ahmed, an eminent journalist of those times, narrated this trend in Muslim politics in his own fashion that „everyone had formed the impression that the political character of the Muslim Legislators was such that whoever formed a ministry the majority would join him‟1 which could be applicable to many of the Hindu members of the House as well. Nazimuddin got full support of the European Group consisting of 25 members in the Assembly. Besides, he had been able to win the confidence of the Anglo-Indians and a section of the Scheduled Caste and a few other members of the Assembly. -

Panel Reduces Fine for Not Wearing Facemask to KD 50 Mps Call to Scrap Kuwait Mosafer App • Jaber Causeway Vaccination Center Opens
SHAWWAL 19, 1442 AH MONDAY, MAY 31, 2021 16 Pages Max 48º Min 29º 150 Fils Established 1961 ISSUE NO: 18447 The First Daily in the Arabian Gulf www.kuwaittimes.net Boris Johnson weds fiancee Hunger stalks India’s poor HK mountaineer on a high after Mexican fans celebrate 5 in ‘secret’ weekend ceremony 7 in pandemic double blow 11 fastest Everest climb by woman 13 return of ‘lucha libre’ Panel reduces fine for not wearing facemask to KD 50 MPs call to scrap Kuwait Mosafer app • Jaber Causeway vaccination center opens By B Izzak questions and even downloading the app to their mobiles. KUWAIT: The National Assembly health commit- He said linking the return of citizens to their tee yesterday agreed to reduce fines for not wear- country with an app that has many technical prob- ing a facemask and abiding by health conditions lems is unacceptable, and accordingly this require- amid the coronavirus pandemic from KD 100 to KD ment must be cancelled and simply replaced with a 50. The reduction came while the committee was PCR test certificate. Saleh said the app must be studying a government-sponsored law proposing scrapped and those responsible for it should be penalties for those who do not comply with health held to account, adding that he has submitted a measures during pandemics. The law must be proposal calling to scrap the app. approved by the Assembly to become effective. MP Osama Al-Shaheen asked Defense Minister A number of lawmakers yesterday strongly criti- Sheikh Hamad Al-Sabah, who is also chairman of cized measures applied by the civil aviation obliging the civil aviation authority, about who designed the travelers to register with the Kuwait Mosafer appli- app and if an aviation company, which is the con- cation and the requirement that travelers must have tractor and main beneficiary of the app, has any link been vaccinated. -

BANGLADESH COUNTRY REPORT April 2004 Country Information & Policy Unit IMMIGRATION & NATIONALITY DIRECTORATE HOME OFFICE
BANGLADESH COUNTRY REPORT April 2004 Country Information & Policy Unit IMMIGRATION & NATIONALITY DIRECTORATE HOME OFFICE, UNITED KINGDOM Bangladesh April 2004 CONTENTS 1. Scope of Document 1.1 - 1.7 2. Geography 2.1 - 2.3 3. Economy 3.1 - 3.3 4. History 4.1 - 4.45 Pre-independence: 1947 – 1971 4.1 - 4.4 1972 –1982 4.5 - 4.8 1983 – 1990 4.9 - 4.14 1991 – 1999 4.15 - 4.26 2000 – the present 4.27 - 4.45 5. State Structures 5.1 - 5.51 The constitution 5.1 - 5.3 - Citizenship and Nationality 5.4 - 5.6 Political System 5.7 - 5.13 Judiciary 5.14 - 5.21 Legal Rights /Detention 5.22 - 5.30 - Death Penalty 5.31 – 5.32 Internal Security 5.33 - 5.34 Prisons and Prison Conditions 5.35 – 5.37 Military Service 5.38 Medical Services 5.39 - 5.45 Educational System 5.46 – 5.51 6. Human Rights 6.1- 6.107 6.A Human Rights Issues 6.1 - 6.53 Overview 6.1 - 6.5 Torture 6.6 - 6.7 Politically-motivated Detentions 6.8 - 6.9 Police and Army Accountability 6.10 - 6.13 Freedom of Speech and the Media 6.14 – 6.23 Freedom of Religion 6.24 - 6.29 Hindus 6.30 – 6.35 Ahmadis 6.36 – 6.39 Christians 6.40 Freedom of Assembly and Association 6.41 Employment Rights 6.42 - 6.47 People Trafficking 6.48 - 6.50 Freedom of Movement 6.51 - 6.52 Authentication of Documents 6.53 6.B Human Rights – Specific Groups 6.54 – 6.85 Ethnic Groups Biharis 6.54 - 6.60 The Tribals of the Chittagong Hill Tracts 6.61 - 6.64 Rohingyas 6.65 – 6.66 Women 6.67 - 6.71 Rape 6.72 - 6.73 Acid Attacks 6.74 Children 6.75 - 6.80 - Child Care Arrangements 6.81 – 6.84 Homosexuals 6.85 Bangladesh April 2004 6.C Human Rights – Other Issues 6.86 – 6.89 Prosecution of 1975 Coup Leaders 6.86 - 6.89 Annex A: Chronology of Events Annex B: Political Organisations Annex C: Prominent People Annex D: References to Source Material Bangladesh April 2004 1. -

Crop Genomics: Present & Future
VI NEXT GENERATION GENOMICS & INTEGRATED BREEDING FOR CROP IMPROVEMENT CONFERENCE ON CROP GENOMICS: PRESENT & FUTURE n Genomics (CE CELEBRATING THE 10TH ANNIVERSARY OF CEG ence i G) cell ANNIVERSARY Ex of (2007-2017) tre en C ICRISAT, HYDERABAD, INDIA CROP GENOMICS: PRESENT & FUTURE CROP GENOMICS: DECEMBER 6-8, 2017 Accelerating Genetic Gains in Crop Breeding Programs throughAccelerating Center Genetic of Excellence Gains in inCrop Genomics Breeding (CEG) Programs throughAccelerating Center Genetic of Excellence Gains in inCrop Genomics Breeding (CEG) Programs through Center of Excellence in Genomics (CEG) Integrated genomics and breeding activities Integrated genomics and breeding activities Integrated genomics and breeding activities Genetic resources Genetic Genetic resources Genetic Genetic resources Genetic Wild species, Landraces, Cultivars, breeding lines, Mapping populations breeding Cultivars, Wild species, Landraces, Superior lines with enhanced tolerance/resistance to abiotic and biotic stress lines, Mapping populations breeding Cultivars, Wild species, Landraces, Superior lines with Wild species, Landraces, Cultivars, breeding lines, Mapping populations breeding Cultivars, Wild species, Landraces, enhanced tolerance/resistance to abiotic and biotic stress Superior lines with Genome sequences enhanced tolerance/resistanceMarker resources to abiotic and biotic stress Genome• Pigeonpea sequences (Nature Biotech 2012, 30:83-89) Marker• Diagnostic resources markers for key traits Genome• PigeonpeaChickpea (Naturesequences (Nature -

The Bridge: Laguardia Community College
President Bill Clinton seen here campaining in New York City for former Mayor, David Dinkin. (photo by Fred Farias) NAFTA PASSED: ATTENTION ALL STUDENTS CLINTON THE CAMPAIGNER PREVAILS * * * * Joseph N. Smith We the members of The Bridge editorial board, would like to By a vote of 234-to-200, Congress passed the North American Free apologize for some of the outdated articles in this issue. The reason for the Trade Agreement that; when it becomes law; many historians may cite as delay of this publication was because of a cosmetic investigation conducted a decision that changed the way America conducts business with its by Dean Lugo Alvarez (Dean of Student Affairs), some Student neighbor to the north, Canada, and to the south, Mexico. The month of Government members and the College Association, in an attempt to November was a pretty good month for President Bill Clinton. Both the punish The Bridge newspaper for publishing an article written by Todd Congress and Senate approved NAFfA and delivered the "Brady Bill". "One" Williams. The situation has been resolved for the moment. Although there is already significant trade between America, Canada and However, while we are able to publish, we will try our best to give you, Mexico, the trade agreement, when signed into law, will turn North the readers, more current and up to date stories in the future. America into the largest free trade zone in the world. At this moment, we are still having some difficulties in trying to get During his campaign for the White House, President Clinton had this issue out to you. -

2011 Project Updates (Incorporating Projects Completed in 2010 and 2009)
Generation Challenge Programme 2011 Project updates (incorporating projects completed in 2010 and 2009) August 2011 Generation Challenge Programme (GCP) Hosted by CIMMYT (Centro Internacional de Mejoramiento de Maíz y Trigo; the International Maize and Wheat Improvement Center) Mailing address: Physical address: c/o CIMMYT, Apdo Postal 6–641 c/o CIMMYT Km 45 Carretera México–Veracruz 06600 Mexico, DF Mexico El Batán, Texcoco, México, CP 56130 Tel: +52 55 5804 2004 Fax: +52 55 5804 7558 Email: [email protected] or [email protected] www.generationcp.org i Published under the Creative Commons Attribution-NonCommercial-Share Alike 2.5 Mexico licence. This licence allows all users to read, copy (in full or in part), distribute or adapt this work, providing that they: (i) acknowledge GCP as the original author of the work; (ii) do not use it for commercial purposes; and, (iii) require other users to observe conditions (i) and ii), (ie, other parties must also acknowledge GCP and use the work for non-commercial purposes). Correct Citation: CGIAR Generation Challenge Programme, 2011. 2011 Project updates (incorporating projects completed in 2010 and 2009). Texcoco, Mexico: Generation Challenge Programme. Compiled by: Gillian Summers Cover illustration: Recomposition based on original artwork by Rhoda Okono entitled Autumn in Africa: revisted Art direction: Miguel Mellado E Editorial note: Project updates are published as presented by the authors, except for updates that extended beyond the set word limit where text beyond this point has been omitted, indicated by continuation dots/suspension points (...) This publication has been printed in black-and-white. For colour images, please contact the appropriate Principal Investigator. -

Bangladesh Annual Report 2013
gets better The splendour and vibrance of life is reverberated exquisitely in nature. Nature epitomizes life and living it well; just as GSK’s brand promise suggests. Hence the cover page of 2013 annual report incorporates our very own elements of nature on a blissful serene backdrop. 52 Directors’ Statement of Responsibilty 53 Auditor’s Report to the Shareholders 54 Statement of Financial Position 55 Income Statement and Statement of Comprehensive Income 56 Statement of Changes in Equity 57 Statement of Cash Flows 58 Notes to the Financial Statements Financials 38 General Information 40 Corporate Governance Frame Work 41 Board of Directors 43 Company Executive Committee 44 Corporate Governance Charter The Company Profile 23 45 Internal Control Framework 24 Product Index 45 Risk Management 28 Product Information 45 Commercial Quality Council 30 Business Activity Details 46 Internal Audit CONTENTS 36 Corporate Social Responsibility Business & Activity Details 46 Salient Feature of Audit Committee Charter 48 Audit Committee Report 49 Responsible Business and Sustainability Development 07 Chairman’s Statement 50 Report on Human Resources Report of Directors to the Shareholders 51 Going Green 10 Corporate Governance 14 Statement of Compliance of Corporate Governance 17 Key Operating and Financial Records 18 Shareholding Information NOTE: 19 Certificate on Compliance of Corporate Governance Brand names appearing throughout this report are trademarks either owned by 20 Analysis of Sales Revenue and/or licensed to GlaxoSmithKline Key Performance