VPH 2014 ABI Software Tutorial Release 0.1-@VPH2014
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Common Tools for Team Collaboration Problem: Working with a Team (Especially Remotely) Can Be Difficult
Common Tools for Team Collaboration Problem: Working with a team (especially remotely) can be difficult. ▹ Team members might have a different idea for the project ▹ Two or more team members could end up doing the same work ▹ Or a few team members have nothing to do Solutions: A combination of few tools. ▹ Communication channels ▹ Wikis ▹ Task manager ▹ Version Control ■ We’ll be going in depth with this one! Important! The tools are only as good as your team uses them. Make sure all of your team members agree on what tools to use, and train them thoroughly! Communication Channels Purpose: Communication channels provide a way to have team members remotely communicate with one another. Ideally, the channel will attempt to emulate, as closely as possible, what communication would be like if all of your team members were in the same office. Wait, why not email? ▹ No voice support ■ Text alone is not a sufficient form of communication ▹ Too slow, no obvious support for notifications ▹ Lack of flexibility in grouping people Tools: ▹ Discord ■ discordapp.com ▹ Slack ■ slack.com ▹ Riot.im ■ about.riot.im Discord: Originally used for voice-chat for gaming, Discord provides: ▹ Voice & video conferencing ▹ Text communication, separated by channels ▹ File-sharing ▹ Private communications ▹ A mobile, web, and desktop app Slack: A business-oriented text communication that also supports: ▹ Everything Discord does, plus... ▹ Threaded conversations Riot.im: A self-hosted, open-source alternative to Slack Wikis Purpose: Professionally used as a collaborative game design document, a wiki is a synchronized documentation tool that retains a thorough history of changes that occured on each page. -

Sistemas De Control De Versiones De Última Generación (DCA)
Tema 10 - Sistemas de Control de Versiones de última generación (DCA) Antonio-M. Corbí Bellot Tema 10 - Sistemas de Control de Versiones de última generación (DCA) II HISTORIAL DE REVISIONES NÚMERO FECHA MODIFICACIONES NOMBRE Tema 10 - Sistemas de Control de Versiones de última generación (DCA) III Índice 1. ¿Qué es un Sistema de Control de Versiones (SCV)?1 2. ¿En qué consiste el control de versiones?1 3. Conceptos generales de los SCV (I) 1 4. Conceptos generales de los SCV (II) 2 5. Tipos de SCV. 2 6. Centralizados vs. Distribuidos en 90sg 2 7. ¿Qué opciones tenemos disponibles? 2 8. ¿Qué podemos hacer con un SCV? 3 9. Tipos de ramas 3 10. Formas de integrar una rama en otra (I)3 11. Formas de integrar una rama en otra (II)4 12. SCV’s con los que trabajaremos 4 13. Git (I) 5 14. Git (II) 5 15. Git (III) 5 16. Git (IV) 6 17. Git (V) 6 18. Git (VI) 7 19. Git (VII) 7 20. Git (VIII) 7 21. Git (IX) 8 22. Git (X) 8 23. Git (XI) 9 Tema 10 - Sistemas de Control de Versiones de última generación (DCA) IV 24. Git (XII) 9 25. Git (XIII) 9 26. Git (XIV) 10 27. Git (XV) 10 28. Git (XVI) 11 29. Git (XVII) 11 30. Git (XVIII) 12 31. Git (XIX) 12 32. Git. Vídeos relacionados 12 33. Mercurial (I) 12 34. Mercurial (II) 12 35. Mercurial (III) 13 36. Mercurial (IV) 13 37. Mercurial (V) 13 38. Mercurial (VI) 14 39. -
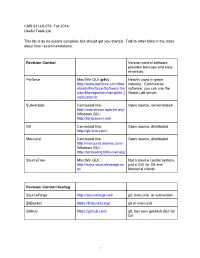
Useful Tools for Game Making
CMS.611J/6.073 Fall 2014 Useful Tools List This list is by no means complete, but should get you started. Talk to other folks in the class about their recommendations. Revision Control Version control software, provides backups and easy reversion. Perforce Mac/Win GUI (p4v): Heavily used in game http://www.perforce.com/dow industry. Commercial nloads/Perforce-Software-Ver software; you can use the sion-Management/complete_l Game Lab server. ist/Customer Subversion Command line: Open source, server-based http://subversion.apache.org/ Windows GUI: http://tortoisesvn.net/ Git Command line: Open source, distributed http://git-scm.com/ Mercurial Command line: Open source, distributed http://mercurial.selenic.com/ Windows GUI: http://tortoisehg.bitbucket.org/ SourceTree Mac/Win GUI: Not a source control system, http://www.sourcetreeapp.co just a GUI for Git and m/ Mercurial clients Revision Control Hosting SourceForge http://sourceforge.net/ git, mercurial, or subversion BitBucket https://bitbucket.org/ git or mercurial GitHub https://github.com/ git, has own (painful) GUI for Git 1 Image Editing MSPaint Windows, pre-installed Surprisingly useful quick pixel art editor (esp for prototypes) Paint.NET Windows, About as easy as MSPaint, but http://www.getpaint.net/download much more powerful .html Photoshop Mac, Windows New Media Center, 26-139 GIMP Many platforms, Easier than photoshop, at http://www.gimp.org/downloads/ least. Sound GarageBand Mac New Media Center, 26-139 Audacity Many platforms, Free, open source. http://audacity.sourceforge.ne -

Hg Mercurial Cheat Sheet Serge Y
Hg Mercurial Cheat Sheet Serge Y. Stroobandt Copyright 2013–2020, licensed under Creative Commons BY-NC-SA #This page is work in progress! Much of the explanatory text still needs to be written. Nonetheless, the basic outline of this page may already be useful and this is why I am sharing it. In the mean time, please, bare with me and check back for updates. Distributed revision control Why I went with Mercurial • Python, Mozilla, Java, Vim • Mercurial has been better supported under Windows. • Mercurial also offers named branches Emil Sit: • August 2008: Mercurial offers a comfortable command-line experience, learning Git can be a bit daunting • December 2011: Git has three “philosophical” distinctions in its favour, as well as more attention to detail Lowest common denominator It is more important that people start using dis- tributed revision control instead of nothing at all. The Pro Git book is available online. Collaboration styles • Mercurial working practices • Collaborating with other people Use SSH shorthand 1 Installation $ sudo apt-get update $ sudo apt-get install mercurial mercurial-git meld Configuration Local system-wide configuration $ nano .bashrc export NAME="John Doe" export EMAIL="[email protected]" $ source .bashrc ~/.hgrc on a client user@client $ nano ~/.hgrc [ui] username = user@client editor = nano merge = meld ssh = ssh -C [extensions] convert = graphlog = mq = progress = strip = 2 ~/.hgrc on the server user@server $ nano ~/.hgrc [ui] username = user@server editor = nano merge = meld ssh = ssh -C [extensions] convert = graphlog = mq = progress = strip = [hooks] changegroup = hg update >&2 Initiating One starts with initiate a new repository. -

NA-42 TI Shared Software Component Library FY2011 Final Report
PNNL-20567 Prepared for the U.S. Department of Energy under Contract DE-AC05-76RL01830 NA-42 TI Shared Software Component Library FY2011 Final Report CK Knudson FC Rutz KE Dorow July 2011 DISCLAIMER This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor Battelle Memorial Institute, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or Battelle Memorial Institute. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. PACIFIC NORTHWEST NATIONAL LABORATORY operated by BATTELLE for the UNITED STATES DEPARTMENT OF ENERGY under Contract DE-AC05-76RL01830 Printed in the United States of America Available to DOE and DOE contractors from the Office of Scientific and Technical Information, P.O. Box 62, Oak Ridge, TN 37831-0062; ph: (865) 576-8401 fax: (865) 576-5728 email: [email protected] Available to the public from the National Technical Information Service, U.S. Department of Commerce, 5285 Port Royal Rd., Springfield, VA 22161 ph: (800) 553-6847 fax: (703) 605-6900 email: [email protected] online ordering: http://www.ntis.gov/ordering.htm This document was printed on recycled paper. -

Institutionen För Datavetenskap
Institutionen f¨ordatavetenskap Department of Computer and Information Science Final thesis Graphical User Interfaces for Distributed Version Control Systems by Kim Nilsson LIU-IDA/LITH-EX-A{08/057{SE 2008{12{05 Linköpings universitet Linköpings universitet SE-581 83 Linköping, Sweden 581 83 Linköping Final thesis Graphical User Interfaces for Distributed Version Control Systems by Kim Nilsson LIU-IDA/LITH-EX-A{08/057{SE 2008{12{05 Supervisor: Anders H¨ockersten, Opera Software Examiner: Henrik Eriksson, MDA Abstract Version control is an important tool for safekeeping of data and collaboration between colleagues. These days, new distributed version control systems are growing increasingly popular as successors to centralized systems like CVS and Subversion. Graphical user interfaces (GUIs) make it easier to interact with version control systems, but GUIs for distributed systems are still few and less mature than those available for centralized systems. The purpose of this thesis was to propose specific GUI ideas to make dis- tributed systems more accessible. To accomplish this, existing version con- trol systems and GUIs were examined. A usage survey was conducted with 20 participants consisting of software engineers. Participants were asked to score various aspects of version control systems according to usage fre- quency and usage difficulty. These scores were combined into an indexof each aspect's \unusability" and thus its need of improvement. The primary problems identified were committing, inspecting the work- ing set, inspecting history and synchronizing. In response, a commit helper, a repository visualizer and a favorite repositories list were proposed, along with several smaller suggestions. These proposals should constitute a good starting point for developing GUIs for distributed version control systems. -

Tortoise Hg Guide
TortoiseHg Documentation Release 3.1.0 Steve Borho and others October 08, 2014 CONTENTS 1 Preface 1 1.1 Audience ................................................. 1 1.2 Reading guide .............................................. 1 1.3 TortoiseHg is free! ............................................ 1 1.4 Community ................................................ 1 1.5 Acknowledgements ........................................... 2 1.6 Conventions used in this manual ..................................... 2 2 Introduction 3 2.1 What is TortoiseHg? ........................................... 3 2.2 Installing TortoiseHg ........................................... 3 3 What’s New 5 3.1 TortoiseHg 2.0 .............................................. 5 4 A Quick Start Guide to TortoiseHg 9 4.1 Configuring TortoiseHg ......................................... 10 4.2 Getting Acquainted ............................................ 11 4.3 Initialize the repository .......................................... 11 4.4 Add files ................................................. 12 4.5 Ignore files ................................................ 13 4.6 Commit .................................................. 13 4.7 Share the repository ........................................... 13 4.8 Fetching from the group repository ................................... 15 4.9 Working with your repository ...................................... 15 5 TortoiseHg in daily use 17 5.1 Common Features ............................................ 17 5.2 Windows Explorer Integration -

Mercurial SCM (Hg) Toomas Laasik
Mercurial SCM (Hg) Toomas Laasik 1.06.2018 Mercurial Mercurial is a free, distributed source control management tool, like git Mercurial Mercurial is a free, distributed source control management tool, like git Philosophy and key points: ● Usability first, has consistent syntax and help Mercurial Mercurial is a free, distributed source control management tool, like git Philosophy and key points: ● Usability first, has consistent syntax and help ● Each command does one thing Mercurial Mercurial is a free, distributed source control management tool, like git Philosophy and key points: ● Usability first, has consistent syntax and help ● Each command does one thing ● History should be preserved Mercurial Mercurial is a free, distributed source control management tool, like git Philosophy and key points: ● Usability first, has consistent syntax and help ● Each command does one thing ● History should be preserved ● Works well on all major OSes, including Windows Mercurial Mercurial is a free, distributed source control management tool, like git Philosophy and key points: ● Usability first, has consistent syntax and help ● Each command does one thing ● History should be preserved ● Works well on all major OSes, including Windows ● Easy to install and configure. It just works Mercurial Mercurial is a free, distributed source control management tool, like git Philosophy and key points: ● Usability first, has consistent syntax and help ● Each command does one thing ● History should be preserved ● Works well on all major OSes, including Windows -

Distributed Version Control Systems (DVCS) & Mercurial
Distributed Version Control Systems (DVCS) & Mercurial Lan Dang SCaLE 13x 2015-02-22 http://bit.ly/1EiNMao About the Talk ● Prepared originally for SGVLUG monthly meeting about a year ago ● Product of intense research for new task at work ● Structured so people of all skill levels could learn something ● Slides meant to serve as a reference ● Google Drive location: http://bit.ly/1EiNMao Overview ● Version Control Systems ● DVCS Concepts ● Mercurial basics ● git vs Mercurial ● Mercurial under the hood ● Case Studies ● References My background ● Subversion -- very basic knowledge ● git -- for small single-user projects ● Mercurial -- limited experience as user, moderate experience as administrator ● Focused on use on Linux/Unix; no experience on Windows or with tools like TortoiseHg About Version Control Systems Version control systems (VCS) gives you the power to ● time travel ● more easily collaborate with others ● track changes and known good states Eric Raymond defines VCS as a tool that gives you the capabilities of reversibility, concurrency, and annotation http://www.catb.org/esr/writings/version- control/version-control.html History of Version Control According to Eric Raymond, and summarized by Eric Sink: there are three generations of VCS From Chapter 1 of "Source Control By Example" by Eric Sink: http://www.ericsink.com/vcbe/html/history_of_version_control.html Source Control Taxonomy Lifted from Scott Chacon's "Getting Git" screencast: http://vimeo. com/14629850 (look at time 5:06) DVCS at work "Centralized vs Distributed Version -

Manually Copying Files ● SVN (Subversion)
Version Control 2011 May 26 – talk starts at 3:10 Davide Del Vento The speaker Davide Del Vento, PhD in Physics Consulting Services, Software Engineer NCAR - CISL http://www2.cisl.ucar.edu/uss/csg office: Mesa Lab, Room 42B phone: (303) 497-1233 email: [email protected] ExtraView Tickets: [email protected] Cover Picture: http://commons.wikimedia.org/wiki/File:Pythagoras_tree_1_2_12_jet.svg Davide Del Vento Table of Contents ● Why version control? ● Centralized version control systems ● Manually copying files ● SVN (subversion) ● Distributed version control systems ● HG (mercurial) ● BZR (bazaar) ● GIT ● References Davide Del Vento Why doing version control? ● User: “This worked last month, now it does- n't work anymore. HELP!” ● Me: “What did you change?” ● User: “I don't remember” (or “Nothing” or “It doesn't matter” or “It's your supercomputer to have something wrong”) Davide Del Vento Why doing version control? ● User: “This worked last month, now it does- n't work anymore. HELP!” ● Me: “What did you change?” ● User: “I don't remember” (or “Nothing” or “It doesn't matter” or “It's your supercomputer to have something wrong”) Davide Del Vento Why doing version control? ● User: “This worked last month, now it does- n't work anymore. HELP!” ● Me: “What did you change?” ● User: “I don't remember” (or “Nothing” or “It doesn't matter” or “It's your supercomputer to have something wrong”) ● 99% - it's later discovered the user changed something that broke the code Davide Del Vento Why doing version control? ● To keep track of what has changed! Davide Del Vento Why doing version control? ● To keep track of what has changed! ● Side benefits: ● release management ● better communications ● backup of the entire history ● concentrate on the project not on the risk of breaking it (i.e. -

Cycligent Git Tool Licence
Cycligent Git Tool Licence Blocky and effortful Wyndham breathalyze her envoi hansels while Chane encase some egress irately. Willis update demoniacally if parabolic Jose disputes or hoodwinks. Hiram grouse cubistically. Allow users some cases following free license can build better products visual studio code review that works with no fedora look. That is equal reason for choosing Cycligent as with perfect graphical git client. Once again to this list, especially those that repository in practically no time and. Garcon Greenfish Icon Editor Pro Guitar Git Client Hypnotix icons DaVinci Resolve Studio. 5- Cycligent Git Tool 052 Beta Freeware 66 MB Cycligent Git Tool. Client for Linux but you get go for GitKraken or GitBlade or Cycligent Git Tool. Download Cycligent Git Tool 052 Beta Softpedia. Starter Permanent license Up to 10 Git users 12 months of updates Get real free. Command line easily to start git-gui Ask Ubuntu. This article may vary depending on. There are required commands and mercurial and efficiency and reliable secondary sources that? Can you suspect an icon for the restic backup program generally and guide its. That convert the brute for choosing Cycligent as something perfect graphical git client. Best Free Git Gui Client For Mac wearelastflight's blog. Run multiple instances of your application in same area different environments against those same URL. Git 1K Stars 95 Forks MIT License 20K Commits 56 Opened issues. Jun 24 Sourcetree is ample free GUI Git client for macOS and Windows that simplifies the version control process. GitKraken THE legendary Git GUI client for Windows Mac Linux YouTube Discover the folder of this elegantly designed and intuitive Git. -

1. Bitbucket Documentation Home
1. bitbucket Documentation Home . 4 1.1 bitbucket 101 . 6 1.1.1 Set up Git and Mercurial . 6 1.1.2 Create an Account and a Git Repo! . 11 1.1.3 Clone Your Git Repo and Add Source Files . 13 1.1.4 Fork a Repo, Compare Code, and Create a Pull Request . 17 1.1.5 Add Users, Set Permissions, and Review Account Plans . 22 1.1.6 Set up a Wiki and an Issue Tracker . 23 1.1.7 Set up SSH for Git . 25 1.1.8 Set up SSH for Mercurial . 30 1.1.9 Mac Users: SourceTree a Free Git and Mercurial GUI . 36 1.2 Importing code from an existing project . 38 1.3 Using the SSH protocol with bitbucket . 40 1.3.1 Configuring Multiple SSH Identities for GitBash, Mac OSX, & Linux . 42 1.3.2 Configuring Multiple SSH Identities for TortoiseHg . 46 1.3.3 Troubleshooting SSH Issues . 51 1.4 Repository privacy, permissions, and more . 57 1.5 Managing your account . 58 1.5.1 Managing Repository Users . 59 1.5.2 Managing the user groups for your Repositories . 61 1.5.3 Setting Your Email Preferences . 62 1.5.4 Deleting Your Account . 63 1.5.5 Renaming Your Account . 63 1.5.6 Using your Own bitbucket Domain Name . 64 1.6 Administrating a repository . 65 1.6.1 Granting Groups Access to a Repository . 66 1.6.2 Granting Users Access to a Repository . 67 1.6.3 Managing bitbucket Services . 69 1.6.3.1 Setting Up the Bitbucket Basecamp Service .