Mitophagy in Human Diseases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
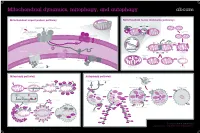
Mitochondrial Dynamics, Mitophagy, and Autophagy
Mitochondrial dynamics, mitophagy, and autophagy Mitochondrial import protein pathway Mitochondrial fusion and fission pathways iΔΨm, (OXPHOSh) Ca2+ Fusion ++++ (Cell differentiationi) N Internal signal β-barrel outer Opa1 SH ER sequence SH membrane precursors Mfn1/2 SH SH GTP GTP C Inner membrane and GTP GTP Outer membrane Matrix carrier precursors 22 70 GTP fusion (N-terminal presequences) 35 GTP 20 37 Tom complex 5 Sam complex Opa1 Cytosol Outer 6 Sam50 7 Mdm10 membrane DRP1 Fis1 Inner membrane fusion Tim8-Tim13 Mim 1 Tom40 Bax/Bak apoptosis Outer membrane HR2 regions S-S Tim9-Tim10 S-S Inner Erv1 95 A 54 mitochondrial S-S Reorganization Mia40 space 21 Fission sequestration 50 18 Tim22 Translocation Depolarization to meet iΔΨm ATP needs Tim23 KREBS Cycle NADH FADH2 17 Oxa1 Tim 22 complex Mia Mba1 Mdm I ATP 44 38 H+ II 17 H+ Inner membrane 16 +++ III ADP - Pi 18 Ribosome Oxa H+ IV mtHsp70 H+ ATP +++ Mge1 MPP Healthy mitochondrion Mitophagy Matrix Tim 23 complex Mitophagy pathway Autophagy pathway Lysosomal hydroiase Lysosome Hypoxia, Nutrient/Growth Rapamycin Starvation condition Ubiquitin Cytosol Parkin factor deprivation Lamp Stress Damaged mitochondrion Parkin Parkin iΔΨm Parkin MARF PINK1 PINK1 PINK1 Mfn1 VDAC1 Parkin AMPK mTOR Mfn2 PINK1 PINK1 Phagophore Autophagosome Autophagolyosome PARL LC3-II PINK1-L PINK1-S Parkin P FIS1 ATG13 ULK LC3-II Membrane PINK1 PINK1 FIP200 PINK1 MIRO2 P Bak PINK1 MIRO1 Parkin Parkin Parkin Cytoplasmic Cytosol proLC3 macromolecule LC3-II Atg4B Atg7/LC3-I Atg5/Atg12/Atg16 Organelle Atg9 PE Atg18 Atg2 -

The Anthelmintic Niclosamide Is a Potent TMEM16A Antagonist That Fully Bronchodilates Airways
bioRxiv preprint doi: https://doi.org/10.1101/254888; this version posted January 27, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. The anthelmintic niclosamide is a potent TMEM16A antagonist that fully bronchodilates airways Kent Miner1, Benxian Liu1, Paul Wang2, Katja Labitzke2,†, Kevin Gaida1, Jian Jeffrey Chen3, Longbin Liu3,†, Anh Leith1,†, Robin Elliott1, Kathryn Henckels1, Esther Trueblood4,†, Kelly Hensley4, Xing-Zhong Xia1, Oliver Homann5, Brian Bennett1, Mike Fiorino1, John Whoriskey1, Sabine Escobar1, Gang Yu1, Joe McGivern2, Min Wong1, Teresa L. Born1,†, Alison Budelsky1,†, Mike Comeau1, Dirk Smith1,†, Jonathan Phillips1, James A. Johnston1, Kerstin Weikl2,†, David 2 1,* 2,†.* 1,† ,* Powers , Deanna Mohn , Andreas Hochheimer , John K. Sullivan 1Department of Inflammation Research, Amgen Inc., Thousand Oaks, CA and Seattle, WA, USA 2Department of Therapeutic Discovery, Amgen Inc., Thousand Oaks, CA, USA and Regensburg, Germany 3Department of Medicinal Chemistry, Amgen Inc., Thousand Oaks, CA, USA 4Department of Comparative Biology and Safety Sciences, Amgen Inc., Seattle, WA, Thousand Oaks, CA and South San Francisco, CA, USA 5Genome Analysis Unit, Amgen Inc., South San Francisco, CA, USA †Present address; see Additional Information section *Corresponding authors. E-mails: [email protected] (J.K.S); [email protected] (A.H.); [email protected] (D.M.) Abstract There is an unmet need in severe asthma where approximately 40% of patients exhibit poor -agonist responsiveness, suffer daily symptoms and show frequent exacerbations. 2+ Antagonists of the Ca -activated-Cl¯ channel, TMEM16A, offers a new mechanism to bronchodilate airways and block the multiple contractiles operating in severe disease. -

Involvement of Impaired Autophagy and Mitophagy in Neuro-2A Cell
www.nature.com/scientificreports OPEN Involvement of impaired autophagy and mitophagy in Neuro-2a cell damage under hypoxic and/or high- Received: 19 May 2017 Accepted: 15 January 2018 glucose conditions Published: xx xx xxxx Yufei Song, Yu Du, Wenying Zou, Yan Luo, Xiaojie Zhang & Jianliang Fu Chronic cerebral hypoperfusion (CCH) plays an insidious role in the development of cognitive impairment. Considerable evidence suggests that Diabetes Mellitus (DM) as a vascular risk factor may exacerbate CCH and is closely related to cognitive decline. Dysregulation of autophagy is known to be associated with the pathogenesis of neurodegenerative diseases such as Alzheimer’s disease. To elucidate the role of autophagy in CCH- and/or DM-related pathogenesis, mouse neuroblastoma Neuro-2a cells were exposed to hypoxia and/or high glucose for 48 h, mimicking CCH complicated with DM pathologies. Chronic hypoxia reduced cell proliferation and increased levels of cleaved caspase-3, whereas high glucose had no obvious synergistic toxic efect. Accumulation of autophagic vacuoles under hypoxia may be due to both autophagy impairment and induction, with the former accounting for Neuro-2a cell death. Additionally, aberrant accumulation of mitochondria in Neuro-2a cells may be attributed to insufcient BNIP3-mediated mitophagy due to poor interaction between BNIP3 and LC3-II. Despite the lack of a signifcant cytotoxic efect of high glucose under our experimental conditions, our data indicated for the frst time that impaired autophagy degradation and inefcient BNIP3-mediated mitophagy may constitute mechanisms underlying neuronal cell damage during chronic hypoxia. Chronic cerebral hypoperfusion (CCH) is a normal process related to ageing that likely contributes to age-related memory loss1. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Endoplasmic Reticulum–Mitochondria Crosstalk in NIX-Mediated Murine Cell Death Abhinav Diwan Washington University School of Medicine in St
Washington University School of Medicine Digital Commons@Becker ICTS Faculty Publications Institute of Clinical and Translational Sciences 2009 Endoplasmic reticulum–mitochondria crosstalk in NIX-mediated murine cell death Abhinav Diwan Washington University School of Medicine in St. Louis Scot J. Matkovich Washington University School of Medicine in St. Louis Qunying Yuan University of Cincinnati Wen Zhao University of Cincinnati Atsuko Yatani University of Medicine and Dentistry of New Jersey See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/icts_facpubs Recommended Citation Diwan, Abhinav; Matkovich, Scot J.; Yuan, Qunying; Zhao, Wen; Yatani, Atsuko; Brown, Joan Heller; Molkentin, Jeffery D.; Kranias, Evangelia G.; and Dorn, Gerald W. II, "Endoplasmic reticulum–mitochondria crosstalk in NIX-mediated murine cell death". Journal of Clinical Investigation, 203-212. 2009. Paper 4. https://digitalcommons.wustl.edu/icts_facpubs/4 This Article is brought to you for free and open access by the Institute of Clinical and Translational Sciences at Digital Commons@Becker. It has been accepted for inclusion in ICTS Faculty Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Abhinav Diwan, Scot J. Matkovich, Qunying Yuan, Wen Zhao, Atsuko Yatani, Joan Heller Brown, Jeffery D. Molkentin, Evangelia G. Kranias, and Gerald W. Dorn II This article is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/icts_facpubs/4 Research article Endoplasmic reticulum–mitochondria crosstalk in NIX-mediated murine cell death Abhinav Diwan,1 Scot J. Matkovich,1 Qunying Yuan,2 Wen Zhao,2 Atsuko Yatani,3 Joan Heller Brown,4 Jeffery D. -

Anthelmintic Activity of Yeast Particle-Encapsulated Terpenes
molecules Article Anthelmintic Activity of Yeast Particle-Encapsulated Terpenes Zeynep Mirza 1, Ernesto R. Soto 1 , Yan Hu 1,2, Thanh-Thanh Nguyen 1, David Koch 1, Raffi V. Aroian 1 and Gary R. Ostroff 1,* 1 Program in Molecular Medicine, University of Massachusetts Medical School, Worcester, MA 01605, USA; [email protected] (Z.M.); [email protected] (E.R.S.); [email protected] (Y.H.); [email protected] (T.-T.N.); [email protected] (D.K.); raffi[email protected] (R.V.A.) 2 Department of Biology, Worcester State University, Worcester, MA 01602, USA * Correspondence: gary.ostroff@umassmed.edu; Tel.: 508-856-1930 Academic Editor: Vaclav Vetvicka Received: 2 June 2020; Accepted: 24 June 2020; Published: 27 June 2020 Abstract: Soil-transmitted nematodes (STN) infect 1–2 billion of the poorest people worldwide. Only benzimidazoles are currently used in mass drug administration, with many instances of reduced activity. Terpenes are a class of compounds with anthelmintic activity. Thymol, a natural monoterpene phenol, was used to help eradicate hookworms in the U.S. South circa 1910. However, the use of terpenes as anthelmintics was discontinued because of adverse side effects associated with high doses and premature stomach absorption. Furthermore, the dose–response activity of specific terpenes against STNs has been understudied. Here we used hollow, porous yeast particles (YPs) to efficiently encapsulate (>95%) high levels of terpenes (52% w/w) and evaluated their anthelmintic activity on hookworms (Ancylostoma ceylanicum), a rodent parasite (Nippostrongylus brasiliensis), and whipworm (Trichuris muris). We identified YP–terpenes that were effective against all three parasites. -

Dimerization of Mitophagy Receptor BNIP3L/NIX Is Essential for Recruitment of Autophagic Machinery
Autophagy ISSN: 1554-8627 (Print) 1554-8635 (Online) Journal homepage: https://www.tandfonline.com/loi/kaup20 Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery Mija Marinković, Matilda Šprung & Ivana Novak To cite this article: Mija Marinković, Matilda Šprung & Ivana Novak (2020): Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery, Autophagy, DOI: 10.1080/15548627.2020.1755120 To link to this article: https://doi.org/10.1080/15548627.2020.1755120 View supplementary material Accepted author version posted online: 14 Apr 2020. Published online: 24 Apr 2020. Submit your article to this journal Article views: 69 View related articles View Crossmark data Full Terms & Conditions of access and use can be found at https://www.tandfonline.com/action/journalInformation?journalCode=kaup20 AUTOPHAGY https://doi.org/10.1080/15548627.2020.1755120 RESEARCH PAPER Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery Mija Marinković a, Matilda Šprung b, and Ivana Novak a aSchool of Medicine, University of Split, Split, Croatia; bFaculty of Science, University of Split, Split, Croatia ABSTRACT ARTICLE HISTORY Mitophagy is a conserved intracellular catabolic process responsible for the selective removal of Received 8 August 2019 dysfunctional or superfluous mitochondria to maintain mitochondrial quality and need in cells. Here, Revised 31 March 2020 we examine the mechanisms of receptor-mediated mitophagy activation, with the focus on BNIP3L/NIX Accepted 2 April 2020 mitophagy receptor, proven to be indispensable for selective removal of mitochondria during the KEYWORDS terminal differentiation of reticulocytes. The molecular mechanisms of selecting damaged mitochondria Autophagy; dimerization; from healthy ones are still very obscure. -

The Role of Translocator Protein TSPO in Hallmarks of Glioblastoma
cancers Review The Role of Translocator Protein TSPO in Hallmarks of Glioblastoma Laura-Marie Ammer 1, Arabel Vollmann-Zwerenz 1, Viktoria Ruf 2, Christian H. Wetzel 3 , Markus J. Riemenschneider 4, Nathalie L. Albert 5, Philipp Beckhove 6 and Peter Hau 1,* 1 Wilhelm Sander-NeuroOncology Unit and Department of Neurology, University Hospital Regensburg, 93053 Regensburg, Germany; [email protected] (L.-M.A.); [email protected] (A.V.-Z.) 2 Center for Neuropathology and Prion Research, Ludwig Maximilians University of Munich, 81377 Munich, Germany; [email protected] 3 Molecular Neurosciences, Department of Psychiatry and Psychotherapy, University of Regensburg, 93053 Regensburg, Germany; [email protected] 4 Department of Neuropathology, Regensburg University Hospital, 93053 Regensburg, Germany; [email protected] 5 Department of Nuclear Medicine, Ludwig-Maximilians-University Munich, 81377 Munich, Germany; [email protected] 6 Regensburg Center for Interventional Immunology (RCI) and Department Internal Medicine III, University Hospital Regensburg, 93053 Regensburg, Germany; [email protected] * Correspondence: [email protected] Received: 14 September 2020; Accepted: 9 October 2020; Published: 14 October 2020 Simple Summary: The translocator protein (TSPO) has been under extensive investigation as a specific marker in positron emission tomography (PET) to visualize brain lesions following injury or disease. In recent years, TSPO is increasingly appreciated as a potential novel therapeutic target in cancer. In Glioblastoma (GBM), the most malignant primary brain tumor, TSPO expression levels are strongly elevated and scientific evidence accumulates, hinting at a pivotal role of TSPO in tumorigenesis and glioma progression. The aim of this review is to summarize the current literature on TSPO with respect to its role both in diagnostics and especially with regard to the critical hallmarks of cancer postulated by Hanahan and Weinberg. -

Mitoq Inhibits Hepatic Stellate Cell Activation and Liver Fibrosis by Enhancing PINK1/Parkin-Mediated Mitophagy
MitoQ Inhibits Hepatic Stellate Cell Activation and Liver Fibrosis by Enhancing PINK1/parkin-mediated Mitophagy Shi-Ying Dou Second Hospital of Hebei Medical University Jiu-Na Zhang Second Hospital of Hebei Medical University Xiao-Li Xie Second Hospital of Hebei Medical University Ting Liu Second Hospital of Hebei Medical University Jun-Li Hu Second Hospital of Hebei Medical University Xiao-Yu Jiang Second Hospital of Hebei Medical University Miao-Miao Wang Second Hospital of Hebei Medical University Hui-Qing Jiang ( [email protected] ) Second Hospital of Hebei Medical University https://orcid.org/0000-0002-5235-1457 Research Article Keywords: Liver brosis, hepatic stellate cell, ubiquinone, PINK1 mitophagy Posted Date: March 30th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-344963/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/18 Abstract Mitophagy plays an important role in the activation of hepatic stellate cells (HSCs). Mitochondria- targeted ubiquinone (MitoQ) is a mitochondria-targeted antioxidant that reduces the production of intracellular reactive oxygen species (ROS). However, its relationship with mitophagy remains unclear. This study evaluated mitophagy during HSC activation and the effects of MitoQ on mitophagy in cell culture and in an animal model of the activation of HSCs. We found that MitoQ reduced the activation of HSCs and alleviated hepatic brosis. While activation of primary HSCs or LX-2 cells was associated with reduced PINK1/parkin-mediated mitophagy, MitoQ reduced intracellular ROS levels, enhanced PINK1/parkin-mediated mitophagy, and inhibited the activation of HSCs. After knocking down the key mitophagy-related protein, PINK1, in LX-2 cells to block mitophagy, MitoQ intervention failed to inhibit HSC activation. -

(TSPO) in Mitochondrial Bioenergetics and Immune Processes
cells Review The Translocator Protein (TSPO) in Mitochondrial Bioenergetics and Immune Processes Calina Betlazar 1,2,* , Ryan J. Middleton 1 , Richard Banati 1,2 and Guo-Jun Liu 1,2,* 1 Human Health, Australian Nuclear Science and Technology Organisation, New Illawarra Road, Lucas Heights, NSW 2234, Australia; [email protected] (R.J.M.); [email protected] (R.B.) 2 Discipline of Medical Imaging & Radiation Sciences, Faculty of Medicine and Health, Brain and Mind Centre, University of Sydney, 94 Mallett Street, Camperdown, NSW 2050, Australia * Correspondence: [email protected] (C.B.); [email protected] (G-J.L.) Received: 11 January 2020; Accepted: 19 February 2020; Published: 24 February 2020 Abstract: The translocator protein (TSPO) is an outer mitochondrial membrane protein that is widely used as a biomarker of neuroinflammation, being markedly upregulated in activated microglia in a range of brain pathologies. Despite its extensive use as a target in molecular imaging studies, the exact cellular functions of this protein remain in question. The long-held view that TSPO plays a fundamental role in the translocation of cholesterol through the mitochondrial membranes, and thus, steroidogenesis, has been disputed by several groups with the advent of TSPO knockout mouse models. Instead, much evidence is emerging that TSPO plays a fundamental role in cellular bioenergetics and associated mitochondrial functions, also part of a greater role in the innate immune processes of microglia. In this review, we examine the more direct experimental literature surrounding the immunomodulatory effects of TSPO. We also review studies which highlight a more central role for TSPO in mitochondrial processes, from energy metabolism, to the propagation of inflammatory responses through reactive oxygen species (ROS) modulation. -

Antiparasitic Properties of Cardiovascular Agents Against Human Intravascular Parasite Schistosoma Mansoni
pharmaceuticals Article Antiparasitic Properties of Cardiovascular Agents against Human Intravascular Parasite Schistosoma mansoni Raquel Porto 1, Ana C. Mengarda 1, Rayssa A. Cajas 1, Maria C. Salvadori 2 , Fernanda S. Teixeira 2 , Daniel D. R. Arcanjo 3 , Abolghasem Siyadatpanah 4, Maria de Lourdes Pereira 5 , Polrat Wilairatana 6,* and Josué de Moraes 1,* 1 Research Center for Neglected Diseases, Guarulhos University, Praça Tereza Cristina 229, São Paulo 07023-070, SP, Brazil; [email protected] (R.P.); [email protected] (A.C.M.); [email protected] (R.A.C.) 2 Institute of Physics, University of São Paulo, São Paulo 05508-060, SP, Brazil; [email protected] (M.C.S.); [email protected] (F.S.T.) 3 Department of Biophysics and Physiology, Federal University of Piaui, Teresina 64049-550, PI, Brazil; [email protected] 4 Ferdows School of Paramedical and Health, Birjand University of Medical Sciences, Birjand 9717853577, Iran; [email protected] 5 CICECO-Aveiro Institute of Materials & Department of Medical Sciences, University of Aveiro, 3810-193 Aveiro, Portugal; [email protected] 6 Department of Clinical Tropical Medicine, Faculty of Tropical Medicine, Mahidol University, Bangkok 10400, Thailand * Correspondence: [email protected] (P.W.); [email protected] (J.d.M.) Citation: Porto, R.; Mengarda, A.C.; Abstract: The intravascular parasitic worm Schistosoma mansoni is a causative agent of schistosomiasis, Cajas, R.A.; Salvadori, M.C.; Teixeira, a disease of great global public health significance. Praziquantel is the only drug available to F.S.; Arcanjo, D.D.R.; Siyadatpanah, treat schistosomiasis and there is an urgent demand for new anthelmintic agents. -

De Novo Neurosteroidogenesis in Human Microglia: Involvement of the 18 Kda Translocator Protein
International Journal of Molecular Sciences Article De novo Neurosteroidogenesis in Human Microglia: Involvement of the 18 kDa Translocator Protein Lorenzo Germelli 1,† , Eleonora Da Pozzo 1,† , Chiara Giacomelli 1 , Chiara Tremolanti 1 , Laura Marchetti 1, Christian H. Wetzel 2 , Elisabetta Barresi 1 , Sabrina Taliani 1 , Federico Da Settimo 1 , Claudia Martini 1,* and Barbara Costa 1 1 Department of Pharmacy, University of Pisa, 56126 Pisa, Italy; [email protected] (L.G.); [email protected] (E.D.P.); [email protected] (C.G.); [email protected] (C.T.); [email protected] (L.M.); [email protected] (E.B.); [email protected] (S.T.); [email protected] (F.D.S.); [email protected] (B.C.) 2 Department of Psychiatry and Psychotherapy, Molecular Neurosciences, University of Regensburg, 93053 Regensburg, Germany; [email protected] * Correspondence: [email protected]; Tel.: +3905-0221-9523 † These authors contributed equally to this work. Abstract: Neuroactive steroids are potent modulators of microglial functions and are capable of counteracting their excessive reactivity. This action has mainly been ascribed to neuroactive steroids released from other sources, as microglia have been defined unable to produce neurosteroids de novo. Unexpectedly, immortalized murine microglia recently exhibited this de novo biosynthesis; herein, de novo neurosteroidogenesis was characterized in immortalized human microglia. The results demonstrated that C20 and HMC3 microglial cells constitutively express members of the Citation: Germelli, L.; Da Pozzo, E.; neurosteroidogenesis multiprotein machinery—in particular, the transduceosome members StAR Giacomelli, C.; Tremolanti, C.; and TSPO, and the enzyme CYP11A1. Moreover, both cell lines produce pregnenolone and transcrip- Marchetti, L.; Wetzel, C.H.; Barresi, E.; tionally express the enzymes involved in neurosteroidogenesis.