Glucose-Mediated N-Glycosylation of SCAP Is Essential for SREBP-1 Activation and Tumor Growth
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Systematic Review on the Implications of O-Linked Glycan Branching and Truncating Enzymes on Cancer Progression and Metastasis
cells Review A Systematic Review on the Implications of O-linked Glycan Branching and Truncating Enzymes on Cancer Progression and Metastasis 1, 1, 1 1,2,3, Rohitesh Gupta y, Frank Leon y, Sanchita Rauth , Surinder K. Batra * and Moorthy P. Ponnusamy 1,2,* 1 Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center, Omaha, NE 68105, USA; [email protected] (R.G.); [email protected] (F.L.); [email protected] (S.R.) 2 Fred and Pamela Buffett Cancer Center, Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE 681980-5900, USA 3 Department of Pathology and Microbiology, UNMC, Omaha, NE 68198-5900, USA * Correspondence: [email protected] (S.K.B.); [email protected] (M.P.P.); Tel.: +402-559-5455 (S.K.B.); +402-559-1170 (M.P.P.); Fax: +402-559-6650 (S.K.B. & M.P.P.) Equal contribution. y Received: 21 January 2020; Accepted: 12 February 2020; Published: 14 February 2020 Abstract: Glycosylation is the most commonly occurring post-translational modifications, and is believed to modify over 50% of all proteins. The process of glycan modification is directed by different glycosyltransferases, depending on the cell in which it is expressed. These small carbohydrate molecules consist of multiple glycan families that facilitate cell–cell interactions, protein interactions, and downstream signaling. An alteration of several types of O-glycan core structures have been implicated in multiple cancers, largely due to differential glycosyltransferase expression or activity. Consequently, aberrant O-linked glycosylation has been extensively demonstrated to affect biological function and protein integrity that directly result in cancer growth and progression of several diseases. -

Glycosylation: Rising Potential for Prostate Cancer Evaluation
cancers Review Glycosylation: Rising Potential for Prostate Cancer Evaluation Anna Kałuza˙ * , Justyna Szczykutowicz and Mirosława Ferens-Sieczkowska Department of Chemistry and Immunochemistry, Wroclaw Medical University, Sklodowskiej-Curie 48/50, 50-369 Wroclaw, Poland; [email protected] (J.S.); [email protected] (M.F.-S.) * Correspondence: [email protected]; Tel.: +48-71-770-30-66 Simple Summary: Aberrant protein glycosylation is a well-known hallmark of cancer and is as- sociated with differential expression of enzymes such as glycosyltransferases and glycosidases. The altered expression of the enzymes triggers cancer cells to produce glycoproteins with specific cancer-related aberrations in glycan structures. Increasing number of data indicate that glycosylation patterns of PSA and other prostate-originated proteins exert a potential to distinguish between benign prostate disease and cancer as well as among different stages of prostate cancer development and aggressiveness. This review summarizes the alterations in glycan sialylation, fucosylation, truncated O-glycans, and LacdiNAc groups outlining their potential applications in non-invasive diagnostic procedures of prostate diseases. Further research is desired to develop more general algorithms exploiting glycobiology data for the improvement of prostate diseases evaluation. Abstract: Prostate cancer is the second most commonly diagnosed cancer among men. Alterations in protein glycosylation are confirmed to be a reliable hallmark of cancer. Prostate-specific antigen is the biomarker that is used most frequently for prostate cancer detection, although its lack of sensitivity and specificity results in many unnecessary biopsies. A wide range of glycosylation alterations in Citation: Kałuza,˙ A.; Szczykutowicz, prostate cancer cells, including increased sialylation and fucosylation, can modify protein function J.; Ferens-Sieczkowska, M. -

Glycosylation of Immune Receptors in Cancer
cells Review Glycosylation of Immune Receptors in Cancer Ruoxuan Sun, Alyssa Min Jung Kim and Seung-Oe Lim * Department of Medicinal Chemistry and Molecular Pharmacology, Purdue Institute of Drug Discovery, Purdue Center for Cancer Research, Purdue University, West Lafayette, IN 47907, USA; [email protected] (R.S.); [email protected] (A.M.J.K.) * Correspondence: [email protected]; Tel.: +1-765-494-3531 Abstract: Evading host immune surveillance is one of the hallmarks of cancer. Immune checkpoint therapy, which aims to eliminate cancer progression by reprogramming the antitumor immune response, currently occupies a solid position in the rapidly expanding arsenal of cancer therapy. As most immune checkpoints are membrane glycoproteins, mounting attention is drawn to asking how protein glycosylation affects immune function. The answers to this fundamental question will stimulate the rational development of future cancer diagnostics and therapeutic strategies. Keywords: glycosylation; N-glycan; immune receptor; immune checkpoint therapy; cancer 1. Introduction The immune system can eliminate malignancy at an early stage by recognizing the antigenic peptide epitope presented by neoplastically transformed cells [1,2]. However, a subset of tumor cells may evolve to coexist with antitumor immunity by a process defined as immunoediting [3]. To combat the immune attack and maintain the balance with the host, cancer cells develop a series of approaches such as (1) downregulation of antigen Citation: Sun, R.; Kim, A.M.J.; Lim, presentation to make them invisible to immunity/the immune system, (2) production S.-O. Glycosylation of Immune of immunomodulatory cytokines to establish an immune suppressive environment, and Receptors in Cancer. Cells 2021, 10, (3) expression of surface immune checkpoint proteins such as programmed cell death 1 1100. -

Design of Glycosylation Sites by Rapid Synthesis and Analysis of Glycosyltransferases
ARTICLES https://doi.org/10.1038/s41589-018-0051-2 Design of glycosylation sites by rapid synthesis and analysis of glycosyltransferases Weston Kightlinger 1,2,7, Liang Lin2,3,7, Madisen Rosztoczy3, Wenhao Li3, Matthew P. DeLisa 4,5, Milan Mrksich 1,2,3,6* and Michael C. Jewett 1,2* Glycosylation is an abundant post-translational modification that is important in disease and biotechnology. Current methods to understand and engineer glycosylation cannot sufficiently explore the vast experimental landscapes required to accurately predict and design glycosylation sites modified by glycosyltransferases. Here we describe a systematic platform for glycosyl- ation sequence characterization and optimization by rapid expression and screening (GlycoSCORES), which combines cell-free protein synthesis and mass spectrometry of self-assembled monolayers. We produced six N- and O-linked polypeptide-modify- ing glycosyltransferases from bacteria and humans in vitro and rigorously determined their substrate specificities using 3,480 unique peptides and 13,903 unique reaction conditions. We then used GlycoSCORES to optimize and design small glycosylation sequence motifs that directed efficient N-linked glycosylation in vitro and in the Escherichia coli cytoplasm for three heterolo- gous proteins, including the human immunoglobulin Fc domain. We find that GlycoSCORES is a broadly applicable method to facilitate fundamental understanding of glycosyltransferases and engineer synthetic glycoproteins. rotein glycosylation is the post-translational attachment of O-linked polypeptide N-acetylgalactosaminyltransferase (ppGalN- oligosaccharides (glycans), most commonly at asparagine AcT), O-linked N-acetylglucosamine transferase (OGT), and oligo- (N-linked) or serine and threonine (O-linked) amino acid side saccharyltransferase (OST) enzyme families. Such enzymes are of P 1,2 chains . -

Native Glycosylation Is Important for Biological Function and Activity of Recombinant Human Proteins
Native Glycosylation Is Important For Biological Function and Activity of Recombinant Human Proteins INTRODUCTION Mark Azam, Ph.D. VP - R&D and Manufacturing Patricia Ahrweiler, Ph.D. Sr. Marketing Consultant SUMMARY Although recombinant human cytokines are commercially available from bacterial, eukaryotic, and mammalian cell expression systems (E. coli, SF9, CHO), human cell- expressed (HEK293) glycoproteins offer the distinct advantage of native human glycosylation. In this Whitepaper, several examples illustrating the importance of correct glycosylation in cytokine activity, stability and half-life, are provided for cytokines widely used in stem cell research. the in to to in the in the in the tion to N to the to the threonine or the to in vivo in E. coli to there in the no or the in Erythropoietin Erythropoietin the in in the other to the EPO (Humankine) glycostructures and the amount of sialic acid in the glycan trees (Croset, 2012). 1 a release of the glycan structures, Calf serum fetuin containing sialylated N and O used as a control for endoglycosidase results that a large of neutral glycans, of ) is mast endothelial acids, sites for glycosylation and cysteine residues forming three 12 and 20 glycosylation. a man cell line, migrates as a of due a a an molecular mass of a due to glycosylation at one glycosylation site and its of is for efficient secretion, the role of glycosylation is unclear. the The bioactivity of VEGF165 was determined by its ability to induce proliferation of human umbilical vein endothelial cells. This supports the importance of correct glycosylation in the biological properties of VEGF165 in cell culture. -

Development of a Whole Cell Fermentation Process for Glycosylation of Small Molecule Flavonoids
Development of a Whole Cell Fermentation Process for Glycosylation of Small Molecule Flavonoids Dissertation Zur Erlangung der Würde des Doktors der Naturwissenschaften des Fachbereichs Biologie, der Fakultät für Mathematik, Informatik und Natur- wissenschaften, der Universität Hamburg vorgelegt von Tanja Vanessa Heyer aus Norderstedt Hamburg 2019 Vollständiger Abdruck der von der Fakultät für Mathematik, Informatik und Natur- wissenschaften der Universität Hamburg zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Gutachter: Prof. Dr. Wolfgang R. Streit PD Dr. Andreas Pommerening-Röser Tag der Disputation: 08.11.2019 Eidesstattliche Versicherung Hiermit erkläre ich an Eides statt, dass ich die vorliegende Dissertationsschrift selbst verfasst und keine anderen als die angegebenen Quellen und Hilfsmittel benutzt habe. ____________________________________ (Tanja Heyer) Funding This work was part of the project “GO-Bio 6: GLYCONIC - Selektiv glykosylierte Polyphenole” funded by the GO-Bio initiative from the German Federal Ministry of Education and Research (BMBF, funding number: 031A376). A b s t r a c t | I Abstract Flavonoids are known to have multiple beneficial effects on human health. These make them desired compounds as bioactives, e.g. in cosmeceutical products. The glycosylation of flavonoids has positive impact on their solubility, bioavailability, and stability and is furthermore able to improve their physiological effects. In this study, flavonoid glycosides were produced in a whole-cell biocatalytic process in Escherichia coli by directed transfer of activated sugar residues to hydroxy groups of the polyphenolic scaffold within an enzymatic reaction by use of glycosyltransferases. The goal of this work was the development and optimization of the production process for glycosylated flavonoids in gram-scale, comprising the upstream (USP) and downstream (DSP) processing. -
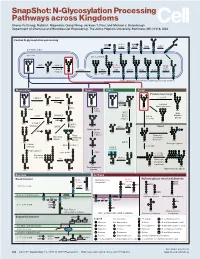
N-Glycosylation Processing Pathways Across Kingdoms Cheng-Yu Chung, Natalia I
SnapShot: N-Glycosylation Processing Pathways across Kingdoms Cheng-Yu Chung, Natalia I. Majewska, Qiong Wang, Jackson T. Paul, and Michael J. Betenbaugh Department of Chemical and Biomolecular Engineering, The Johns Hopkins University, Baltimore, MD 21218, USA Central N-glycosylation processing UDP ALG13 GDP ALG7 ALG14 ALG1 ALG2 ALG11 P P P P P CYTOPLASM P P P P P P GOLGI ER LUMEN GNTI α -Man IA En Bloc ALG3 Transfer ALG9 α-Man IB ER Man I α -Glc I OST ALG10 ALG8 ALG6 ALG12 Asn α -Man IC Asn Asn Asn α -Glc II P P P P P Asn P P P P P ALGs: asparagine-linked N-glycosylation processing enzymes Mammalian Insect Plant Fungi Filamentous fungi α-Man II FUT8 α-1,2-MT XYLT GDP Terminal Asn Asn Asn Asn UDP Asn Galactofuranose GNTII Asn Asn FUT8 f ± α-Gal Asn FUTC3 GNTII High FUT11 α -GALT B14GALTI Och1p Mannose FUT12 glycan α-Man II α-1,2-MT UDP Asn Asn ST3GAL4 Asn Asn GNTIV ST6GAL1 GNTIII GNTV Asn GNase Yeast Hybrid glycan P CMP GNase Asn Asn Tetra- α-Man II Mannosyl- Bisecting antennary phosphate glycan glycan transferase Asn Asn Asn Asn GALT1 Asn ST8SIA2 Core type glycan GNTs B14GALTI α-1,6-MT ST8SIA4 Asn Lewis A structure Polysialylation α-Man PolyLacNAc α-1,2-MT Alternative α-1,3-MT P sialic acid FUT13 ] [ STs Mannosyl- n phosphate Asn Asn Asn transferase Asn Asn Paucimannose Hypermannose glycan Asn Asn Asn glycans Bacteria Archaea Archaea glycan structural diversity Block transfer En Bloc Methanococcus En Bloc Transfer Transfer maripaludis OCH3 Thr OCH AgIB Thr OCH3 3 PERIPLASM PglB B B Asn P - P Asn OSO3 P Asn Asn INNER MEMBRANE P Asn P -

TBV25H Expressed in Yeast and Purified Using Nickel-NTA Agarose
© 1994 Nature Publishing Group http://www.nature.com/naturebiotechnology /RESEARCH• Production, Purification and lmmunogenicity of a Malaria Transmission-Blocking Vaccine Candidate: TBV25H Expressed in Yeast and Purified Using Nickel-NTA Agarose David C. Kaslow* and Joseph Shlloach 1 Molecular Vaccine Section, Laboratory of Malaria Research, National Institute of Allergy and Infectious Diseases. 'Biotechnology Unit, Laboratory of Cellular and Developmental Biology, National Insitute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD 20892. *Corresponding author (e-mail: [email protected]). We have constructed a second generation malaria transmission-blocking vaccine candidate based on Pfs25, the predominate surface protein of Plasmodium falciparum zygotes, to overcome potential produc tion problems with the original construct. Four modifications were made: (1) addition of the last cysteine residue of the fourth epidermal growth factor like-domain of Pfs25; (2) mutagenesis of asparagine-linked glycosylation sites with glutamine rather than alanine; (3) addition of a six histidine tag at the carboxy terminus for highly efficient purification ofrecombinant protein on nickel-NTA agarose; and (4) fermenta tion that combines continuous glucose fed-batch methodology with pH-controlled glucose addition and a terminal ethanol feed. The resulting product, TBV25H (Transmission-Blocking Vaccine based on Pfs25 with a Histidine tag), appears to be a more potent antigen and immunogen than the original construct, and the fermentation and -

Glycosylation of Amino Acids and Efficient Synthesis of Glycosphingolipids
Georgia State University ScholarWorks @ Georgia State University Chemistry Theses Department of Chemistry 8-11-2015 Glycosylation of Amino Acids and Efficient Synthesis of Glycosphingolipids Jonathan Wooten Georgia State University, [email protected] Follow this and additional works at: https://scholarworks.gsu.edu/chemistry_theses Recommended Citation Wooten, Jonathan, "Glycosylation of Amino Acids and Efficient Synthesis of Glycosphingolipids." Thesis, Georgia State University, 2015. https://scholarworks.gsu.edu/chemistry_theses/74 This Thesis is brought to you for free and open access by the Department of Chemistry at ScholarWorks @ Georgia State University. It has been accepted for inclusion in Chemistry Theses by an authorized administrator of ScholarWorks @ Georgia State University. For more information, please contact [email protected]. GLYCOSYLATION OF AMINO ACIDS AND EFFICIENT SYNTHESIS OF GLYCOSPHINGOLIPIDS by JONATHAN VALENTINE WOOTEN Under the Direction of Peng George Wang, PhD ABSTRACT Glycoscience is an emerging field of science that focuses on the study of the structure, biosynthesis, biology, and evolution of saccharides (sugars). It covers a broad range of subjects including microwave-assisted synthesis as well as sphingolipid synthesis. In this field, knowledge is limited due to the complexity of carbohydrates and their derivatives. Therefore, it is all the more important that synthesis of these complex molecules occurs in order to fully understand their biological significance. The following report summarizes -

Role of Protein Glycosylation in Immune Regulation Ann Rheum Dis: First Published As 10.1136/Ard.52.Suppl 1.S22 on 1 March 1993
S22 Annals of the Rheumatic Diseases 1993; 52: S22-S29 Role of protein glycosylation in immune regulation Ann Rheum Dis: first published as 10.1136/ard.52.Suppl_1.S22 on 1 March 1993. Downloaded from Elizabeth F Hounsell, Michael J Davies Most cell surface and secreted proteins are those of the outer branches of N-linked glycosylated - that is, they have one or more glycosylation (although there are differences). oligosaccharide chains covalently attached to In both chains the diversity of oligosaccharide specific amino acids. These oligosaccharides sequences is manifest in the large number of usually make up a significant amount of the ways the monosaccharides can be linked hydrodynamic mass of the molecules and also together (that is, through 1-4 hydroxyl groups, have effects on protein conformation, surface with and without branching, in (x or 3 expression, stability, circulating half life, configuration, etc). Each linkage has a activity, and antigenicity. In addition, oligo- particular set of allowed conformations in saccharide sequences are themselves solution which provide specific orientations of recognised as antigens and as ligands for functional groups for molecular recognition. carbohydrate binding proteins (lectins). The Such epitopes usually extend over a tri- to immune system exemplifies the diversity of heptasaccharide sequence, but topographical oligosaccharide structure and function, which epitopes caused by folding back of long this review will aim to communicate. In carbohydrate chains onto themselves or onto particular, the relevance of oligosaccharide protein are possible. In addition, the oligo- heterogeneity, antigenicity, and immune saccharide cores of 0-linked chains are in close regulatory activity in autoimmunity and association with the protein backbone, which microbial pathogenesis will be considered. -

UDP-Glucose Pyrophosphorylase 2, a Regulator of Glycogen Synthesis and Glycosylation, Is Critical for Pancreatic Cancer Growth
UDP-glucose pyrophosphorylase 2, a regulator of glycogen synthesis and glycosylation, is critical for pancreatic cancer growth Andrew L. Wolfea, Qingwen Zhoub, Eneda Toskac, Jacqueline Galeasa, Angel A. Kua,d, Richard P. Kochee, Sourav Bandyopadhyaya,d, Maurizio Scaltritic,f, Carlito B. Lebrillab,g,h, Frank McCormicka,i,1,2, and Sung Eun Kima,1,2,3,4 aHelen Diller Comprehensive Cancer Center, University of California, San Francisco, CA 94158; bDepartment of Chemistry, University of California, Davis, CA 95616; cHuman Oncology and Pathogenesis Program, Memorial Sloan Kettering Cancer Center, New York, NY 10065; dBioengineering and Therapeutic Sciences, University of California, San Francisco, CA 94158; eCenter for Epigenetics Research, Memorial Sloan Kettering Cancer Center, New York, NY 10065; fDepartment of Pathology, Memorial Sloan Kettering Cancer Center, New York, NY 10065; gDepartment of Biochemistry and Molecular Medicine, University of California, Davis, CA 95616; hFoods for Health Institute, University of California, Davis, CA 95616; and iNational Cancer Institute RAS Initiative, Cancer Research Technology Program, Frederick National Laboratory for Cancer Research, Frederick, MD 21701 Contributed by Frank McCormick, June 28, 2021 (sent for review February 26, 2021; reviewed by Adrian L. Harris and Alec Kimmelman) UDP-glucose pyrophosphorylase 2 (UGP2), the enzyme that synthe- We further characterize two regulatory functions of UGP2 in sizes uridine diphosphate (UDP)-glucose, rests at the convergence of PDAC cells: first, that -

Chemistry and Biology of Asparagine-Linked Glycosylation*
Pure Appl. Chem., Vol. 71, No. 5, pp. 777±787, 1999. Printed in Great Britain. q 1999 IUPAC Chemistry and biology of asparagine-linked glycosylation* Barbara Imperiali,² Sarah E. O'Connor, Tamara Hendrickson and Christine Kellenberger Division of Chemistry and Chemical Engineering,California Institute of Technology, Pasadena, CA 91125, USA Abstract: The biosynthesis of glycoprotein conjugates is a complex process that involves the collective action of numerous enzymes. Recent research on the chemistry and biology of asparagine-linked glycosylation in our group has been focused on two speci®c areas. These are the development of potent inhibitors of oligosaccharyl transferase and the investigation of the conformational consequences of the glycosylation process. Since asparagine-linked glycosylation is an essential eukaryotic process, an understanding of the details of this complex transformation is of utmost importance both to fundamental biochemistry and to a considera- tion of the mechanisms of homeostatic control. INTRODUCTION Protein glycosylation impacts both the functional capacity and structural framework of all glycoproteins [1,2]. The carbohydrate modi®cations of proteins fall into three general categories: N-linked modi®cation of asparagine [3±5], O-linked modi®cation of serine or threonine [6], and glycosylphosphatidyl inositol derivatization of the C-terminus carboxyl group [7]. Each of these transformations is catalyzed by one or more enzymes which demonstrate different peptide sequence requirements and reaction speci®cities. This paper will focus on N-linked glycosylation, which is the most common of the eukaryotic glycosylation reactions [4]. N-linked glycosylation is catalyzed by a single enzyme, oligosaccharyl transferase (OT), and involves the co-translational transfer of a lipid-linked tetradecasaccharide (GlcNAc2-Man9-Glc3) to an asparagine side chain (in the consensus sequence Asn-Xaa-Ser/Thr) within a nascent polypeptide.