Truxima Prescribing Information
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

FTSE Korea 30/18 Capped
2 FTSE Russell Publications 19 August 2021 FTSE Korea 30/18 Capped Indicative Index Weight Data as at Closing on 30 June 2021 Index weight Index weight Index weight Constituent Country Constituent Country Constituent Country (%) (%) (%) Alteogen 0.19 KOREA Hyundai Engineering & Construction 0.35 KOREA NH Investment & Securities 0.14 KOREA AmoreG 0.15 KOREA Hyundai Glovis 0.32 KOREA NHN 0.07 KOREA Amorepacific Corp 0.65 KOREA Hyundai Heavy Industries 0.29 KOREA Nong Shim 0.08 KOREA Amorepacific Pfd. 0.08 KOREA Hyundai Marine & Fire Insurance 0.13 KOREA OCI 0.17 KOREA BGF Retail 0.09 KOREA Hyundai Merchant Marine 1.02 KOREA Orion 0.21 KOREA BNK Financial Group 0.18 KOREA Hyundai Mipo Dockyard 0.15 KOREA Ottogi 0.06 KOREA Celltrion Healthcare 0.68 KOREA Hyundai Mobis 1.53 KOREA Paradise 0.07 KOREA Celltrion Inc 2.29 KOREA Hyundai Motor 2.74 KOREA Posco 1.85 KOREA Celltrion Pharm 0.24 KOREA Hyundai Motor 2nd Pfd. 0.33 KOREA Posco Chemical 0.32 KOREA Cheil Worldwide 0.14 KOREA Hyundai Motor Pfd. 0.21 KOREA Posco International 0.09 KOREA CJ Cheiljedang 0.3 KOREA Hyundai Steel 0.33 KOREA S1 Corporation 0.13 KOREA CJ CheilJedang Pfd. 0.02 KOREA Hyundai Wia 0.13 KOREA Samsung Biologics 0.92 KOREA CJ Corp 0.11 KOREA Industrial Bank of Korea 0.22 KOREA Samsung C&T 0.94 KOREA CJ ENM 0.15 KOREA Kakao 3.65 KOREA Samsung Card 0.08 KOREA CJ Logistics 0.12 KOREA Kangwon Land 0.23 KOREA Samsung Electro-Mechanics 0.81 KOREA Coway 0.36 KOREA KB Financial Group 1.78 KOREA Samsung Electronics 25.36 KOREA Daewoo Engineering & Construction 0.12 KOREA KCC Corp 0.12 KOREA Samsung Electronics Pfd. -
Holdings-Report.Pdf
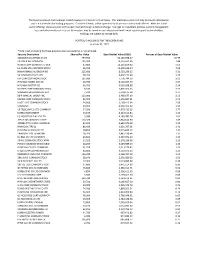
The Fund is a closed-end exchange traded management Investment company. This material is presented only to provide information and is not intended for trading purposes. Closed-end funds, unlike open-end funds are not continuously offered. After the initial public offering, shares are sold on the open market through a stock exchange. Changes to investment policies, current management fees, and other matters of interest to investors may be found in each closed-end fund's most recent report to shareholders. Holdings are subject to change daily. PORTFOLIO HOLDINGS FOR THE KOREA FUND as of July 31, 2021 *Note: Cash (including for these purposes cash equivalents) is not included. Security Description Shares/Par Value Base Market Value (USD) Percent of Base Market Value SAMSUNG ELECTRONICS CO 793,950 54,183,938.27 20.99 SK HYNIX INC COMMON 197,500 19,316,452.95 7.48 NAVER CORP COMMON STOCK 37,800 14,245,859.60 5.52 LG CHEM LTD COMMON STOCK 15,450 11,309,628.34 4.38 HANA FINANCIAL GROUP INC 225,900 8,533,236.25 3.31 SK INNOVATION CO LTD 38,200 8,402,173.44 3.26 KIA CORP COMMON STOCK 107,000 7,776,744.19 3.01 HYUNDAI MOBIS CO LTD 26,450 6,128,167.79 2.37 HYUNDAI MOTOR CO 66,700 6,030,688.98 2.34 NCSOFT CORP COMMON STOCK 8,100 5,802,564.66 2.25 SAMSUNG BIOLOGICS CO LTD 7,230 5,594,175.18 2.17 KB FINANCIAL GROUP INC 123,000 5,485,677.03 2.13 KAKAO CORP COMMON STOCK 42,700 5,456,987.61 2.11 HUGEL INC COMMON STOCK 24,900 5,169,415.34 2.00 SAMSUNG 29,900 4,990,915.02 1.93 SK TELECOM CO LTD COMMON 17,500 4,579,439.25 1.77 KOREA INVESTMENT 53,100 4,427,115.84 -

Korea Morning Focus
December 23, 2019 Korea Morning Focus No English report summaries this morning Major Indices Close Chg Chg (%) KOSPI 2,204.18 7.62 0.35 KOSPI 200 295.31 1.00 0.34 KOSDAQ 649.73 1.88 0.29 Turnover ('000 shares, Wbn) Volume Value KOSPI 637,639 5,021 KOSPI 200 75,360 3,446 KOSDAQ 929,806 4,198 Market Cap (Wbn) Value KOSPI 1,481,267 KOSDAQ 234,817 KOSPI Turnover (Wbn) Buy Sell Net Foreign 2,004 1,848 156 Institutional 986 847 139 Retail 1,961 2,283 -322 KOSDAQ Turnover (Wbn) Buy Sell Net Foreign 518 418 100 Institutional 172 164 8 Retail 3,484 3,568 -84 Program Buy / Sell (Wbn) Buy Sell Net KOSPI 1,813 1,611 203 KOSDAQ 485 406 79 Advances & Declines Advances Declines Unchanged KOSPI 437 366 105 KOSDAQ 609 601 134 KOSPI Top 5 Most Active Stocks by Value (Wbn) Price (W) Chg (W) Value Samsung Electronics 56,000 0 676 Hynix 95,000 1,400 263 KODEX Leverage 14,145 65 183 Jooyon 804 31 161 KODEX Kosdaq150 8,005 85 150 Leverage KOSDAQ Top 5 Most Active Stocks by Value (Wbn) Price (W) Chg (W) Value HLB 102,200 3,300 205 Energy Solutions 25,850 2,750 170 BRIDGE 54,300 -8,100 140 BIOTHERAPEUTICS SyntekaBio 15,100 650 113 ESA 2,060 -635 103 Note: As of December 20, 2019 This document is a summary of a report prepared by Mirae Asset Daewoo Co., Ltd. (“Mirae Asset Daewoo”) and published on our website. -

Assessment Report on Extension(S) of Marketing Authorisation
19 September 2019 EMA/CHMP/548703/2019 Committee for Medicinal Products for Human Use (CHMP) Assessment Report on extension(s) of marketing authorisation Remsima International non-proprietary name: infliximab Procedure No. EMEA/H/C/002576/X/0062 Note Assessment report as adopted by the CHMP with all information of a commercially confidential nature deleted. Official address Domenico Scarlattilaan 6 ● 1083 HS Amsterdam ● The Netherlands Address for visits and deliveries Refer to www.ema.europa.eu/how-to-find-us Send us a question Go to www.ema.europa.eu/contact Telephone +31 (0)88 781 6000 An agency of the European Union © European Medicines Agency, 2019. Reproduction is authorised provided the source is acknowledged. Table of contents 1. Background information on the procedure ............................................ 11 1.1. Submission of the dossier .................................................................................... 11 1.2. Steps taken for the assessment of the product ....................................................... 11 2. Scientific discussion .............................................................................. 12 2.1. Problem statement ............................................................................................. 12 2.2. About the product .............................................................................................. 13 2.3. Type of Application and aspects on development .................................................... 14 2.4. Quality aspects ................................................................................................. -

Truxima (Rituximab-Abbs) – New and Expanded Indications
Truxima® (rituximab-abbs) – New and expanded indications • On May 23, 2019, the FDA approved Teva and Celltrion’s Truxima (rituximab-abbs), for the treatment of adult patients with previously untreated diffuse large B-cell, CD20-positive non-Hodgkin’s lymphoma (NHL) in combination with (cyclophosphamide, doxorubicin, vincristine, and prednisone) (CHOP) or other anthracycline-based chemotherapy regimens; and with previously untreated and previously treated CD20-positive chronic lymphocytic leukemia (CLL) in combination with fludarabine and cyclophosphamide (FC). • Previously, Truxima was approved for the following: — Relapsed or refractory, low grade or follicular, CD20-positive B-cell NHL as a single agent — Previously untreated follicular, CD20-positive, B-cell NHL in combination with first line chemotherapy and, in patients achieving a complete or partial response to a rituximab product in combination with chemotherapy, as single-agent maintenance therapy — Non-progressing (including stable disease), low-grade, CD20-positive, B-cell NHL as a single agent after first-line cyclophosphamide, vincristine, and prednisone chemotherapy. • Truxima is a biosimilar to Genentech and Biogen’s Rituxan® (rituximab). — Rituxan is also approved for the treatment of adult patients with rheumatoid arthritis, granulomatosis with polyangiitis (Wegener's granulomatosis) and microscopic polyangiitis, and pemphigus vulgaris. • Similar to Rituxan, Truxima carries a boxed warning for fatal infusion-related reactions, severe mucocutaneous reactions, hepatitis B virus reactivation and progressive multifocal leukoencephalopathy. • The most common adverse reactions (≥ 25%) with Truxima use in NHL were infusion-related reactions, fever, lymphopenia, chills, infection and asthenia. • The most common adverse reactions (≥ 25%) with Truxima use in CLL were infusion-related reactions and neutropenia. • The recommended dose of Truxima in the expanded NHL indication is 375 mg/m2 as an intravenous infusion on day 1 of each cycle of chemotherapy, for up to 8 infusions. -
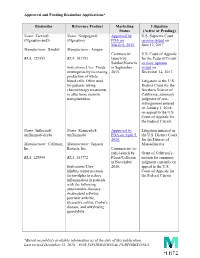
Approved and Pending Biosimilar Applications* *Based on Publicly
Approved and Pending Biosimilar Applications* Biosimilar Reference Product Marketing Litigation Status (Active or Pending) Name: Zarxio® Name: Neupogen® Approved by U.S. Supreme Court (filgrastim-sndz) (filgrastim) FDA on opinion issued on March 6, 2015. June 12, 2017. Manufacturer: Sandoz Manufacturer: Amgen Commercial U.S. Court of Appeals BLA: 125553 BLA: 103353 launch by for the Federal Circuit Sandoz/Novartis en banc opinion Indications/Uses: Treats in September issued on neutropenia by increasing 2015. December 14, 2017. production of white blood cells. Often used Litigation in the U.S. for patients taking District Court for the chemotherapy treatments Northern District of or after bone marrow California; summary transplantation. judgment of non- infringement entered on January 1, 2018; on appeal to the U.S. Court of Appeals for the Federal Circuit. Name: Inflectra® Name: Remicade® Approved by Litigation initiated in (infliximab-dyyb) (infliximab) FDA on April 5, the U.S. District Court 2016. for the District of Manufacturer: Celltrion, Manufacturer: Janssen Massachusetts. Inc. Biotech, Inc. Commercial (at- risk) launch by Grant of Celltrion’s BLA: 125544 BLA: 103772 Pfizer/Celltrion motion for summary in November judgment currently on Indications/Uses: 2016. appeal to the U.S. Inhibits tumor necrosis Court of Appeals for factor-alpha to reduce the Federal Circuit. inflammation in patients with the following autoimmune diseases: rheumatoid arthritis, psoriatic arthritis, ulcerative colitis, Crohn’s disease, and ankylosing spondylitis. *Based on publicly available information as of the date of this publication. Last revised December 13, 2018 – FOR INFORMATIONAL PURPOSES ONLY. Approved and Pending Biosimilar Applications* Name: Erelzi® Name: Enbrel® Approved by Litigation in the U.S. -

CELLTRION Inc. CT-P13 3.5
CELLTRION Inc. CT-P13 3.5 A Randomized, Parallel-Group, Phase I/III Study to Evaluate Efficacy, Pharmacokinetics and Safety between Subcutaneous CT-P13 and Intravenous CT-P13 in Patients with Active Rheumatoid Arthritis 24th March 2019 Statistical Analysis Plan Part 2 – Final Version 4.0 Prepared by: XXXXXXXXX XXXXXXXXXXXXXXXXXXX XXXXXXXXXXXX XXXX XXXXXXX Prepared by: ____________________________ Date: ____/____/____ XXXXXXXXXXXX XXXXXXXX XXXXXXXXXX Approved by: ____________________________ Date: ____/____/____ XXXXXXXXXXXX XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX XXXXXXXXXX Upon review of this document, including table, listing and figure shells, the undersigned approves the final statistical analysis plan. The analysis methods and data presentation are acceptable, and the table, listing and figure production can begin. I Confidential Confidential CELLTRION, Inc. Statistical Analysis Plan, Part 2 – Final Version 4.0 CT-P13 3.5 Date Issued: 24th March 2019 TABLE OF CONTENTS 1. ADMINISTRATIVE STRUCTURE .......................................................................... 6 2. INTRODUCTION........................................................................................................ 6 3. STUDY OBJECTIVE .................................................................................................. 7 3.1. Primary Objective .................................................................................................. 7 3.2. Secondary Objectives............................................................................................ -
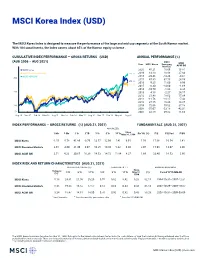
MSCI Korea Index (USD)
MSCI Korea Index (USD) The MSCI Korea Index is designed to measure the performance of the large and mid cap segments of the South Korean market. With 104 constituents, the index covers about 85% of the Korean equity universe . CUMULATIVE INDEX PERFORMANCE — GROSS RETURNS (USD) ANNUAL PERFORMANCE (%) (AUG 2006 – AUG 2021) MSCI Year MSCI Korea Emerging MSCI Markets ACWI IMI MSCI Korea 2020 45.21 18.69 16.81 MSCI Emerging Markets 330.11 2019 13.10 18.88 27.04 MSCI ACWI IMI 2018 -20.46 -14.24 -9.61 300 2017 47.80 37.75 24.58 275.40 2016 9.25 11.60 8.96 252.73 2015 -6.30 -14.60 -1.68 2014 -10.70 -1.82 4.36 200 2013 4.18 -2.27 24.17 2012 21.48 18.63 17.04 2011 -11.76 -18.17 -7.43 2010 27.15 19.20 14.87 100 2009 72.06 79.02 37.18 2008 -55.07 -53.18 -42.01 50 2007 32.58 39.82 11.66 Aug 06 Nov 07 Feb 09 May 10 Aug 11 Nov 12 Feb 14 May 15 Aug 16 Nov 17 Feb 19 May 20 Aug 21 INDEX PERFORMANCE — GROSS RETURNS (%) (AUG 31, 2021) FUNDAMENTALS (AUG 31, 2021) ANNUALIZED Since 1 Mo 3 Mo 1 Yr YTD 3 Yr 5 Yr 10 Yr May 31, 1994 Div Yld (%) P/E P/E Fwd P/BV MSCI Korea -1.55 -5.78 41.80 -0.79 12.57 12.96 7.41 6.91 1.56 17.06 10.54 1.41 MSCI Emerging Markets 2.65 -4.00 21.49 3.07 10.25 10.80 5.22 6.30 2.07 15.98 13.07 2.00 MSCI ACWI IMI 2.51 4.33 30.65 16.38 14.53 14.72 11.84 8.27 1.69 23.40 18.53 2.90 INDEX RISK AND RETURN CHARACTERISTICS (AUG 31, 2021) ANNUALIZED STD DEV (%) 2 SHARPE RATIO 2 , 3 MAXIMUM DRAWDOWN Turnover Since 1 3 Yr 5 Yr 10 Yr 3 Yr 5 Yr 10 Yr May 31, (%) Period YYYY-MM-DD (%) 1994 MSCI Korea 5.18 24.61 21.00 20.20 0.55 0.62 0.42 0.28 82.15 1994-10-31—1997-12-31 MSCI Emerging Markets 8.45 19.23 16.52 17.57 0.54 0.63 0.34 0.43 65.14 2007-10-29—2008-10-27 MSCI ACWI IMI 3.24 18.47 14.81 14.00 0.76 0.92 0.82 0.43 58.28 2007-10-31—2009-03-09 1 Last 12 months 2 Based on monthly gross returns data 3 Based on ICE LIBOR 1M The MSCI Korea Index was launched on Mar 31, 1989. -

Korea Morning Focus
May 23, 2019 Korea Morning Focus Company News & Analysis Major Indices Close Chg Chg (%) LG Electronics (066570/Buy/TP: W99,500) KOSPI 2,064.86 3.61 0.18 5G presents new opportunities KOSPI 200 266.65 0.53 0.20 KOSDAQ 706.93 2.95 0.42 Turnover ('000 shares, Wbn) Volume Value KOSPI 500,633 4,638 KOSPI 200 60,374 2,652 KOSDAQ 712,110 4,222 Market Cap (Wbn) Value KOSPI 1,368,593 KOSDAQ 242,884 KOSPI Turnover (Wbn) Buy Sell Net Foreign 1,300 1,294 5 Institutional 940 1,032 -92 Retail 2,369 2,296 73 KOSDAQ Turnover (Wbn) Buy Sell Net Foreign 458 515 -57 Institutional 231 213 17 Retail 3,505 3,464 41 Program Buy / Sell (Wbn) Buy Sell Net KOSPI 1,056 1,113 -57 KOSDAQ 398 429 -31 Advances & Declines Advances Declines Unchanged KOSPI 390 428 77 KOSDAQ 661 532 89 KOSPI Top 5 Most Active Stocks by Value (Wbn) Price (W) Chg (W) Value Samsung Electronics 43,500 350 463 KODEX Leverage 11,720 30 303 KODEX 200 Futures 7,860 -20 244 Inverse 2X Hynix 68,900 -1,400 213 KODEX Kosdaq150 10,485 40 141 Leverage KOSDAQ Top 5 Most Active Stocks by Value (Wbn) Price (W) Chg (W) Value Hyundai IT 18,300 3,000 367 Nexon GT 17,650 650 188 TPLEX 3,150 420 114 ABL Bio 33,250 2,350 100 Kukil Paper 3,980 110 84 Note: As of May 22, 2019 This document is a summary of a report prepared by Mirae Asset Daewoo Co., Ltd. -

OPERS Letter in Support of Celltrion's Biologics License Application for A
Ohio Public Employees Retirement System February 3, 2016 Stephanie Begansky, PharmD Center for Drug Evaluation and Research Food and Drug Administration 10903 New Hampshire Avenue, WO31-2417 Silver Spring, MD 20933-0002 To the Food and Drug Administration Arthritis Advisory Committee: The Ohio Public Employees Retirement System (“OPERS”) appreciates the opportunity to submit comments to the Food and Drug Administration (“FDA”) Arthritis Advisory Committee regarding the approval of Celltrion Inc.’s Biologics License Application (“BLA”) 125544 for CT-P13, an infliximab biosimilar to Janssen Biotech’s REMICADE (“infliximab”). Upon learning that the FDA had accepted Celltrion’s BLA for a biosimilar infliximab, we felt that it was appropriate to offer our support. OPERS has 222,000 non-Medicare and Medicare Retiree Health Care Program participants, and ensuring that costs within the health care system remain affordable and sustainable is of paramount concern to us. The availability and accessibility of safe, effective, and affordable biosimilar medications is an integral part of OPERS’ long-term strategy to manage its future prescription drug costs for our retiree health care plan and plan participants. Celltrion has submitted significant evidence demonstrating that its infliximab is biosimilar to the US-licensed reference product (Janssen Biotech’s REMICADE); including comparative clinical trials: (1) in combination with methotrexate, reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function -

Morning Focus
August 22, 2018 Korea Morning Focus Company News & Analysis Major Indices Close Chg Chg (%) Shinsegae International (031430/Buy/TP: W260,000) KOSPI 2,270.06 22.18 0.99 Power of platform KOSPI 200 291.93 3.32 1.15 KOSDAQ 787.15 17.37 2.26 Turnover ('000 shares, Wbn) Volume Value KOSPI 290,503 5,093 KOSPI 200 78,660 3,894 KOSDAQ 504,193 3,420 Market Cap (Wbn) Value KOSPI 1,518,199 KOSDAQ 262,063 KOSPI Turnover (Wbn) Buy Sell Net Foreign 1,506 1,400 106 Institutional 1,232 1,249 -17 Retail 2,282 2,433 -151 KOSDAQ Turnover (Wbn) Buy Sell Net Foreign 404 292 111 Institutional 304 167 137 Retail 2,718 2,953 -235 Program Buy / Sell (Wbn) Buy Sell Net KOSPI 1,280 1,225 54 KOSDAQ 250 121 129 Advances & Declines Advances Declines Unchanged KOSPI 523 293 82 KOSDAQ 891 249 102 KOSPI Top 5 Most Active Stocks by Value (Wbn) Price (W) Chg (W) Value Hynix 78,400 3,700 429 Samsung Electronics 44,800 950 422 KODEX KOSDAQ150 15,620 990 247 LEVERAGE SEMCO 143,500 10,000 176 Celltrion 268,500 7,500 167 KOSDAQ Top 5 Most Active Stocks by Value (Wbn) Price (W) Chg (W) Value SillaJen 62,900 2,400 105 ECOMAISTER 13,600 1,450 94 Keayeast 3,335 -40 92 ViroMed 217,200 16,200 83 Daea TI 9,170 50 81 Note: As of August 21, 2018 Mirae Asset Daewoo Research This document is a summary of a report prepared by Mirae Asset Daewoo Co., Ltd. -

INFLECTRA Safely and Effectively
HIGHLIGHTS OF PRESCRIBING INFORMATION 5 mg/kg at 0, 2 and 6 weeks, then every 8 weeks. Some adult patients who These highlights do not include all the information needed to use initially respond to treatment may benefit from increasing the dose to 10 INFLECTRA safely and effectively. See full prescribing information for mg/kg if they later lose their response. INFLECTRA. Pediatric Crohn’s Disease (2.2) 5 mg/kg at 0, 2 and 6 weeks, then every 8 weeks. INFLECTRA (infliximab-dyyb) for Injection, for Intravenous Use Ulcerative Colitis (2.3) Initial U.S. Approval: 2016 5 mg/kg at 0, 2 and 6 weeks, then every 8 weeks. INFLECTRA (infliximab-dyyb) is biosimilar* to REMICADE (infliximab) for Rheumatoid Arthritis (2.4) the indications listed. (1) In conjunction with methotrexate, 3 mg/kg at 0, 2 and 6 weeks, then every 8 WARNING: SERIOUS INFECTIONS and MALIGNANCY weeks. Some patients may benefit from increasing the dose up to 10 mg/kg or See full prescribing information for complete boxed warning treating as often as every 4 weeks. Ankylosing Spondylitis (2.5) Increased risk of serious infections leading to hospitalization or death, 5 mg/kg at 0, 2 and 6 weeks, then every 6 weeks. including tuberculosis (TB), bacterial sepsis, invasive fungal infections Psoriatic Arthritis (2.6) and Plaque Psoriasis (2.7) (such as histoplasmosis) and infections due to other opportunistic 5 mg/kg at 0, 2 and 6 weeks, then every 8 weeks. pathogens. (5.1) Discontinue INFLECTRA if a patient develops a serious infection. (5.1) -----------------------DOSAGE FORMS AND STRENGTHS----------------------- For injection: 100 mg of lyophilized infliximab-dyyb in a 20 mL vial for Perform test for latent TB; if positive, start treatment for TB prior to intravenous infusion.