Loss of the Smallest Subunit of Cytochrome C Oxidase, COX8A, Causes Leigh-Like Syndrome and Epilepsy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity Michael Y. Zhang,1 Siobán B. Keel,2 Tom Walsh,3 Ming K. Lee,3 Suleyman Gulsuner,3 Amanda C. Watts,3 Colin C. Pritchard,4 Stephen J. Salipante,4 Michael R. Jeng,5 Inga Hofmann,6 David A. Williams,6,7 Mark D. Fleming,8 Janis L. Abkowitz,2 Mary-Claire King,3 and Akiko Shimamura1,9,10 M.Y.Z. and S.B.K. contributed equally to this work. 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA; 2Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 3Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; 4Department of Laboratory Medicine, University of Washington, Seattle, WA; 5Department of Pediatrics, Stanford Uni- versity School of Medicine, Stanford, CA; 6Division of Hematology/Oncology, Boston Children’s Hospital, Dana Farber Cancer Insti- tute, and Harvard Medical School, Boston, MA; 7Harvard Stem Cell Institute, Boston, MA; 8Department of Pathology, Boston Children’s Hospital, MA; 9Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 10Department of Pedi- atrics, University of Washington, Seattle, WA, USA ©2014 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2014.113456 Manuscript received on July 22, 2014. Manuscript accepted on September 15, 2014. Correspondence: [email protected] Supplementary Methods Genomics. Libraries were prepared in 96-well format with a Bravo liquid handling robot (Agilent Technologies). One to two micrograms of genomic DNA were sheared to a peak size of 150 bp using a Covaris E series instrument. -

Proteomic Analysis of the Role of the Quality Control Protease LONP1 in Mitochondrial Protein Aggregation
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.12.439502; this version posted April 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Proteomic analysis of the role of the quality control protease LONP1 in mitochondrial protein aggregation Karen Pollecker1, Marc Sylvester2 and Wolfgang Voos1,* 1Institute of Biochemistry and Molecular Biology (IBMB), University of Bonn, Faculty of Medicine, Nussallee 11, 53115 Bonn, Germany 2Core facility for mass spectrometry, Institute of Biochemistry and Molecular Biology (IBMB), University of Bonn, Faculty of Medicine, Nussallee 11, 53115 Bonn, Germany *Corresponding author Email: [email protected] Phone: +49-228-732426 Abbreviations: AAA+, ATPases associated with a wide variety of cellular activities; Δψ, mitochondrial membrane potential; gKD, genetic knockdown; HSP, heat shock protein; m, mature form; mt, mitochondrial; p, precursor form; PQC, protein quality control; qMS, quantitative mass spectrometry; ROS, reactive oxygen species; SILAC, stable isotope labeling with amino acids in cell culture; siRNA, small interfering RNA; TIM, preprotein translocase complex of the inner membrane; TMRE, tetramethylrhodamine; TOM, preprotein translocase complex of the outer membrane; UPRmt, mitochondrial unfolded protein response; WT, wild type. bioRxiv preprint doi: https://doi.org/10.1101/2021.04.12.439502; this version posted April 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -
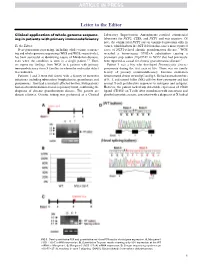
Clinical Application of Whole-Genome Sequencing in Patients with Primary
Letter to the Editor Clinical application of whole-genome sequenc- Laboratory Improvement Amendments–certified commercial ing in patients with primary immunodeficiency laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen examined mutations only in To the Editor: exon 2, which harbors the 2GT deletion that causes most reported Next-generation sequencing, including whole-exome sequenc- cases of NCF1-related chronic granulomatous disease.4 WGS ing and whole-genome sequencing (WES and WGS, respectively), revealed a homozygous 579G>A substitution causing a has been successful at identifying causes of Mendelian diseases, premature stop codon (Trp193X) in NCF1 that had previously even when the condition is seen in a single patient.1-3 Here, been reported as causal for chronic granulomatous disease.5 we report our findings from WGS in 6 patients with primary Patient 3 was a boy who developed Pneumocystis jiroveci immunodeficiency from 5 families in whom the molecular defect pneumonia during the first year of life. There was no family was unknown. history of primary immunodeficiency. Immune evaluation Patients 1 and 2 were full sisters with a history of recurrent demonstrated absent serum IgG and IgA. He had normal numbers infections, including tuberculous lymphadenitis, granulomas, and of B, T, and natural killer (NK) cells by flow cytometry and had pneumonias. They had a similarly affected brother. Both patients normal T-cell proliferative responses to mitogens and antigens. had an absent rhodamine-based respiratory burst, confirming the However, the patient lacked any detectable expression of CD40 diagnosis of chronic granulomatous disease. The parents are ligand (CD154) on T cells after stimulation with ionomycin and distant relatives. -

In Silico Tools for Splicing Defect Prediction: a Survey from the Viewpoint of End Users
© American College of Medical Genetics and Genomics REVIEW In silico tools for splicing defect prediction: a survey from the viewpoint of end users Xueqiu Jian, MPH1, Eric Boerwinkle, PhD1,2 and Xiaoming Liu, PhD1 RNA splicing is the process during which introns are excised and informaticians in relevant areas who are working on huge data sets exons are spliced. The precise recognition of splicing signals is critical may also benefit from this review. Specifically, we focus on those tools to this process, and mutations affecting splicing comprise a consider- whose primary goal is to predict the impact of mutations within the able proportion of genetic disease etiology. Analysis of RNA samples 5′ and 3′ splicing consensus regions: the algorithms used by different from the patient is the most straightforward and reliable method to tools as well as their major advantages and disadvantages are briefly detect splicing defects. However, currently, the technical limitation introduced; the formats of their input and output are summarized; prohibits its use in routine clinical practice. In silico tools that predict and the interpretation, evaluation, and prospection are also discussed. potential consequences of splicing mutations may be useful in daily Genet Med advance online publication 21 November 2013 diagnostic activities. In this review, we provide medical geneticists with some basic insights into some of the most popular in silico tools Key Words: bioinformatics; end user; in silico prediction tool; for splicing defect prediction, from the viewpoint of end users. Bio- medical genetics; splicing consensus region; splicing mutation INTRODUCTION TO PRE-mRNA SPLICING AND small nuclear ribonucleoproteins and more than 150 proteins, MUTATIONS AFFECTING SPLICING serine/arginine-rich (SR) proteins, heterogeneous nuclear ribo- Sixty years ago, the milestone discovery of the double-helix nucleoproteins, and the regulatory complex (Figure 1). -

COX4I2 Antibody (Aa31-80) Rabbit Polyclonal Antibody Catalog # ALS15898
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 COX4I2 Antibody (aa31-80) Rabbit Polyclonal Antibody Catalog # ALS15898 Specification COX4I2 Antibody (aa31-80) - Product Information Application IHC, IF, WB Primary Accession Q96KJ9 Reactivity Human, Mouse, Rat Host Rabbit Clonality Polyclonal Calculated MW 20kDa KDa COX4I2 Antibody (aa31-80) - Additional Information Gene ID 84701 Anti-COX4I2 antibody IHC staining of human kidney. Other Names Cytochrome c oxidase subunit 4 isoform 2, mitochondrial, Cytochrome c oxidase subunit IV isoform 2, COX IV-2, COX4I2, COX4L2 Target/Specificity COX42 Antibody detects endogenous levels Immunofluorescence of COS7 cells, using of total COX42 protein. COX42 Antibody. Reconstitution & Storage Store at -20°C for up to one year. Precautions COX4I2 Antibody (aa31-80) is for research use only and not for use in diagnostic or therapeutic procedures. COX4I2 Antibody (aa31-80) - Protein Information Western blot of extracts from K562 cells, Name COX4I2 treated with insulin 0.01U/ml 15', using COX42 Antibody. Synonyms COX4L2 Function COX4I2 Antibody (aa31-80) - Background Component of the cytochrome c oxidase, the last enzyme in the mitochondrial This protein is one of the nuclear-coded electron transport chain which drives polypeptide chains of cytochrome c oxidase, oxidative phosphorylation. The respiratory the terminal oxidase in mitochondrial electron chain contains 3 multisubunit complexes transport. Page 1/2 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 succinate dehydrogenase (complex II, CII), COX4I2 Antibody (aa31-80) - References ubiquinol- cytochrome c oxidoreductase (cytochrome b-c1 complex, complex III, CIII) Huettemann M.,et al.Gene and cytochrome c oxidase (complex IV, 267:111-123(2001). -

Mutation in the COX4I1 Gene Is Associated with Short Stature, Poor Weight Gain and Increased Chromosomal Breaks, Simulating Fanconi Anemia
European Journal of Human Genetics (2017) 25, 1142–1146 & 2017 Macmillan Publishers Limited, part of Springer Nature. All rights reserved 1018-4813/17 www.nature.com/ejhg ARTICLE Mutation in the COX4I1 gene is associated with short stature, poor weight gain and increased chromosomal breaks, simulating Fanconi anemia Bassam Abu-Libdeh*,1,4, Liza Douiev2,3,4, Sarah Amro1, Maher Shahrour1, Asaf Ta-Shma2, Chaya Miller2,3, Orly Elpeleg2 and Ann Saada*,2,3 We describe a novel autosomal recessive form of mitochondrial disease in a child with short stature, poor weight gain, and mild dysmorphic features with highly suspected Fanconi anemia due to a mutation in COX4I1 gene. Whole Exome Sequencing was performed then followed by Sanger confirmation, identified a K101N mutation in COX4I1, segregating with the disease. This nuclear gene encodes the common isoform of cytochrome c oxidase (COX) subunit 4 (COX 4-1), an integral regulatory part of COX (respiratory chain complex IV) the terminal electron acceptor of the mitochondrial respiratory chain. The patient’s fibroblasts disclosed decreased COX activity, impaired ATP production, elevated ROS production, decreased expression of COX4I1 mRNA and undetectable (COX4) protein. COX activity and ATP production were restored by lentiviral transfection with the wild-type gene. Our results demonstrate the first human mutation in the COX4I1 gene linked to diseases and confirm its role in the pathogenesis. Thus COX4I1 mutations should be considered in any patient with features suggestive of this diagnosis. European Journal of Human Genetics (2017) 25, 1142–1146; doi:10.1038/ejhg.2017.112; published online 2 August 2017 INTRODUCTION normal vaginal delivery with birth weight of 2.8 kg after an uneventful Mitochondrial cytochrome c oxidase (COX, complex IV) is the terminal pregnancy. -

Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients Siobán B. Keel, 1* Angela Scott, 2,3,4 * Marilyn Sanchez-Bonilla, 5 Phoenix A. Ho, 2,3,4 Suleyman Gulsuner, 6 Colin C. Pritchard, 7 Janis L. Abkowitz, 1 Mary-Claire King, 6 Tom Walsh, 6** and Akiko Shimamura 5** 1Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 2Clinical Research Division, Fred Hutchinson Can - cer Research Center, Seattle, WA; 3Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 4Department of Pedi - atrics, University of Washington, Seattle, WA; 5Boston Children’s Hospital, Dana Farber Cancer Institute, and Harvard Medical School, MA; 6Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; and 7Department of Laboratory Medicine, University of Washington, Seattle, WA, USA *SBK and ASc contributed equally to this work **TW and ASh are co-senior authors ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2016.149476 Received: May 16, 2016. Accepted: July 13, 2016. Pre-published: July 14, 2016. Correspondence: [email protected] or [email protected] Supplementary materials Supplementary methods Retrospective chart review Patient data were collected from medical records by two investigators blinded to the results of genetic testing. The following information was collected: date of birth, transplant, death, and last follow-up, -

Loss of COX4I1 Leads to Combined Respiratory Chain Deficiency And
cells Article Loss of COX4I1 Leads to Combined Respiratory Chain Deficiency and Impaired Mitochondrial Protein Synthesis Kristýna Cunˇ átová 1,2, David Pajuelo Reguera 1 , Marek Vrbacký 1, Erika Fernández-Vizarra 3 , Shujing Ding 3, Ian M. Fearnley 3, Massimo Zeviani 3 , Josef Houštˇek 1 , Tomáš Mráˇcek 1,* and Petr Pecina 1,* 1 Laboratory of Bioenergetics, Institute of Physiology, Czech Academy of Sciences, 142 00 Prague, Czech Republic; [email protected] (K.C.);ˇ [email protected] (D.P.R.); [email protected] (M.V.); [email protected] (J.H.) 2 Department of Cell Biology, Faculty of Science, Charles University, 128 00 Prague, Czech Republic 3 MRC Mitochondrial Biology Unit, University of Cambridge, Cambridge CB2 0XY, UK; [email protected] (E.F.-V.); [email protected] (S.D.); [email protected] (I.M.F.); [email protected] (M.Z.) * Correspondence: [email protected] (T.M.); [email protected] (P.P.) Abstract: The oxidative phosphorylation (OXPHOS) system localized in the inner mitochondrial membrane secures production of the majority of ATP in mammalian organisms. Individual OXPHOS complexes form supramolecular assemblies termed supercomplexes. The complexes are linked not only by their function but also by interdependency of individual complex biogenesis or maintenance. For instance, cytochrome c oxidase (cIV) or cytochrome bc1 complex (cIII) deficiencies affect the level of fully assembled NADH dehydrogenase (cI) in monomeric as well as supercomplex forms. It was hypothesized that cI is affected at the level of enzyme assembly as well as at the level of cI stability and maintenance. -

COX6B1 Antibody - N-Terminal Region Rabbit Polyclonal Antibody Catalog # AI15391
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 COX6B1 Antibody - N-terminal region Rabbit Polyclonal Antibody Catalog # AI15391 Specification COX6B1 Antibody - N-terminal region - Product Information Application WB Primary Accession P14854 Other Accession NM_001863, NP_001854 Reactivity Human, Mouse, Rat, Rabbit, Pig, Horse, Yeast, Bovine, Guinea Pig, Dog Predicted Human, Mouse, Host: Rabbit Rat, Rabbit, Pig, Target Name: COX6B1 Horse, Yeast, Sample Tissue: OVCAR-3 Whole cell lysate Bovine, Guinea Pig, Dog s Host Rabbit Antibody Dilution: 1.0μg/ml Clonality Polyclonal Calculated MW 10kDa KDa COX6B1 Antibody - N-terminal region - References COX6B1 Antibody - N-terminal region - Additional Information Taanman J.-W.,et al.Nucleic Acids Res. 17:1766-1766(1989). Gene ID 1340 Taanman J.-W.,et al.Gene 93:285-291(1990). Carrero-Valenzuela R.D.,et al.Gene Alias Symbol COX6B, COXG, 102:229-236(1991). COXVIb1 Ota T.,et al.Nat. Genet. 36:40-45(2004). Other Names Kalnine N.,et al.Submitted (MAY-2003) to the Cytochrome c oxidase subunit 6B1, EMBL/GenBank/DDBJ databases. Cytochrome c oxidase subunit VIb isoform 1, COX VIb-1, COX6B1, COX6B Format Liquid. Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and 2% sucrose. Reconstitution & Storage Add 50 ul of distilled water. Final anti-COX6B1 antibody concentration is 1 mg/ml in PBS buffer with 2% sucrose. For longer periods of storage, store at 20°C. Avoid repeat freeze-thaw cycles. Precautions COX6B1 Antibody - N-terminal region is for Page 1/3 10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 research use only and not for use in diagnostic or therapeutic procedures. -

Structure of the Intact 14-Subunit Human Cytochrome C Oxidase
www.nature.com/cr www.cell-research.com ARTICLE Structure of the intact 14-subunit human cytochrome c oxidase Shuai Zong 1, Meng Wu1, Jinke Gu1, Tianya Liu1, Runyu Guo1 and Maojun Yang1,2 Respiration is one of the most basic features of living organisms, and the electron transport chain complexes are probably the most complicated protein system in mitochondria. Complex-IV is the terminal enzyme of the electron transport chain, existing either as randomly scattered complexes or as a component of supercomplexes. NDUFA4 was previously assumed as a subunit of complex-I, but recent biochemical data suggested it may be a subunit of complex-IV. However, no structural evidence supporting this notion was available till now. Here we obtained the 3.3 Å resolution structure of complex-IV derived from the human supercomplex I1III2IV1 and assigned the NDUFA4 subunit into complex-IV. Intriguingly, NDUFA4 lies exactly at the dimeric interface observed in previously reported crystal structures of complex-IV homodimer which would preclude complex- IV dimerization. Combining previous structural and biochemical data shown by us and other groups, we propose that the intact complex-IV is a monomer containing 14 subunits. Cell Research (2018) 28:1026–1034; https://doi.org/10.1038/s41422-018-0071-1 INTRODUCTION states under physiological conditions, either being assembled into Mitochondria are critical to many cellular activities. Respiration is supercomplexes or freely scattered on mitochondrial inner the central function of mitochondria, and is exquisitely regulated membrane.36 NDUFA4 was originally considered as a subunit of in multiple ways in response to varying cell conditions.1–3 The Complex-I37 but was proposed to belong to Complex-IV recently.38 assembly of respiratory chain complexes, Complex I–IV (CI, NADH: However, the precise location of NDUFA4 in CIV remains unknown, ubiquinone oxidoreductase; CII, succinate:ubiquinone oxidoreduc- and thus this notion still lacks structural support. -

Hypoxia Inducible Factors Modulate Mitochondrial Oxygen Consumption and Transcriptional Regulation of Nuclear-Encoded Electron T
Article pubs.acs.org/biochemistry Hypoxia Inducible Factors Modulate Mitochondrial Oxygen Consumption and Transcriptional Regulation of Nuclear-Encoded Electron Transport Chain Genes † § † ‡ ∥ † † ‡ † Hye Jin Hwang, , Scott G. Lynn, , , Ajith Vengellur, Yogesh Saini, , Elizabeth A. Grier, † § † § ‡ Shelagh M. Ferguson-Miller, , and John J. LaPres*, , , † ‡ § Department of Biochemistry and Molecular Biology, Center for Integrative Toxicology, and Center for Mitochondrial Science and Medicine, Michigan State University, East Lansing, Michigan 48824-1319, United States *S Supporting Information ABSTRACT: Hypoxia inducible factor-1 (HIF1) is a stress-responsive nuclear transcription factor that is activated with a decrease in oxygen availability. HIF1 regulates the expression of genes involved in a cell’s adaptation to hypoxic stress, including those with mitochondrial specific function. To gain a more comprehensive understanding of the role of HIF1 in mitochondrial homeostasis, we studied the link between hypoxia, HIF1 transactivation, and electron transport chain (ETC) function. We established immortalized mouse embryonic fibroblasts (MEFs) for HIF1α wild-type (WT) and null cells and tested whether HIF1α regulates mitochondrial respiration by modulating gene expressions of nuclear-encoded ETC components. High- throughput quantitative real-time polymerase chain reaction was performed to screen nuclear- encoded mitochondrial genes related to the ETC to identify those whose regulation was HIF1α- dependent. Our data suggest that HIF1α regulates transcription of cytochrome c oxidase (CcO) heart/muscle isoform 7a1 (Cox7a1) under hypoxia, where it is induced 1.5−2.5-fold, whereas Cox4i2 hypoxic induction was HIF1α-independent. We propose that adaptation to hypoxic stress of CcO as the main cellular oxygen consumer is mediated by induction of hypoxia-sensitive tissue-specific isoforms. -

Anti-COX7B Antibody
03/19 Anti-COX7B Antibody CATALOG NO.: A1756-100 100 μl. BACKGROUND DESCRIPTION: Cytochrome c oxidase (COX), the terminal component of the mitochondrial respiratory chain, catalyzes the electron transfer from reduced cytochrome c to oxygen. This component is a heteromeric complex consisting of 3 catalytic subunits encoded by mitochondrial genes and multiple structural subunits encoded by nuclear genes. The mitochondrial- encoded subunits function in electron transfer, and the nuclear-encoded subunits may function in the regulation and assembly of the complex. This nuclear gene encodes subunit VIIb, which is highly similar to bovine COX VIIb protein and is found in all tissues. This gene may have several pseudogenes on chromosomes 1, 2, 20 and 22. ALTERNATE NAMES: Cytochrome c oxidase polypeptide VIIb, Cytochrome c oxidase subunit 7B, Cytochrome c oxidase subunit 7B mitochondrial, APLCC. ANTIBODY TYPE: Polyclonal CONCENTRATION: 0.3 mg/ml HOST/ISOTYPE: Rabbit / IgG. IMMUNOGEN: Recombinant protein of human COX7B. MOLECULAR WEIGHT: 9 kDa. PURIFICATION: Affinity purification. FORM: Liquid. FORMULATION: PBS with 0.05% sodium azide, 50% glycerol, PH7.3 SPECIES REACTIVITY: Rat. Human. STORAGE CONDITIONS: Store at -20°C. Avoid freeze / thaw cycles. APPLICATIONS AND USAGE: WB 1:500-1:2000, IHC 1:50-1:200. : IHC staining of paraffin-embedded Human colon cancer using Anti-COX7B Antibody. 155 S. Milpitas Blvd., Milpitas, CA 95035 USA | T: (408)493-1800 F: (408)493-1801 | www.biovision.com | [email protected] 03/19 : Western Blot analysis of 293T cell using Anti-COX7B Antibody. RELATED PRODUCTS: Cox-3 Antibody (3687). Cyclooxygenase (COX) Activity Assay Kit (Fluorometric) (K549). TFPI Antibody (3379).