Shape Data Analysis Using Qushape
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

M-Explorer User's Guide
Table of Contents M-Explorer User’s Guide Table of Contents M-Explorer User’s Guide..............................................1 Chapter 1 Using This Guide.................................................................1-1 Introduction...................................................................................................... 1-1 Key Concepts................................................................................................... 1-2 Chapter Organization .....................................................................................................1-2 Online Help ....................................................................................................................1-2 Manual Conventions ......................................................................................................1-2 Chapter 2 Introduction to M-Explorer .................................................2-1 Introduction...................................................................................................... 2-1 Key Concepts................................................................................................... 2-2 System Component Interaction......................................................................................2-2 OPC Data Access Servers.............................................................................................2-2 M-Inspector ....................................................................................................................2-2 M-Password ...................................................................................................................2-3 -

Iwork '08 Getting Started (Manual)
Overview of iWork Tools All three iWork applications share many of the same tools. The Toolbar and Format Bar At the top of each application window, the toolbar provides controls for common tasks. Each toolbar is described in detail in the appropriate chapter in this book. You can customize the toolbar so that it contains the tools you use most often. To customize the toolbar: m Choose View > Customize Toolbar. The toolbar at the top of each window provides controls for common tasks. The Format Bar provides additional formatting tools. The Format Bar provides quick access to commonly used tools for formatting objects. If the Format Bar isn’t visible beneath the toolbar, click View in the toolbar and choose Show Format Bar to show it. 16 Preface Welcome to iWork ’08 The Inspector Window You can format all elements of your document using the panes of the Inspector window. The Inspector panes are described in detail in the user’s guides. To open the Inspector window: m Click Inspector (a blue i) in the toolbar. Click the buttons along the top to see the different Inspector panes. You can have more than one Inspector window open at a time. To open another Inspector window: m Choose View > New Inspector, or Option-click one of the buttons at the top of the Inspector window. Preface Welcome to iWork ’08 17 To see what a control does, rest the pointer over it until its help tag appears. The Media Browser This window provides quick access to all the files in your iTunes library, your iPhoto library, your Aperture library, and your Movies folder. -
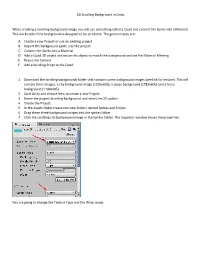
2D Scrolling Background in Unity When Creating a Scrolling
2D Scrolling Background in Unity When creating a scrolling background image you will use something called a Quad and convert the Sprite into a Material. This works only if the background is designed to be scrollable. The general steps are: A. Create a new Project or use an existing project. B. Import the background sprite into the project C. Convert the Sprite into a Material D. Add a Quad 3D object and resize this object to match the background and set the Material Filtering E. Resize the Camera F. Add a Scrolling Script to the Quad 1. Download the scrolling backgrounds folder that contains some background images (see link for lecture). This will contain three images, a city background image (1230x600), a space background (1782x600) and a trees background (1100x405) 2. Start Unity and choose New to create a new Project. 3. Name the project Scrolling Background and select the 2D option. 4. Create the Project. 5. In the Assets folder create two new folders named Sprites and Scripts. 6. Drag these three background images into the sprites folder. 7. Click the scrolling city background image in the Sprites folder. The Inspector window shows the properties: You are going to change the Texture Type and the Wrap mode. 8. Set the Texture Type to Default and the Wrap Mode to Repeat. 9. Scroll down in the Inspector window and click the Apply button. 10. Repeat these steps for the other two sprites. 11. Click the Game Object menu, choose 3D, and choose Quad: You want to resize the quad object so it matches the background. -

BD Cellquest™ Pro Analysis Tutorial
BD CellQuest™ Pro Analysis Tutorial This tutorial guides you through an analysis example using BD CellQuest Pro software. If you are already familiar with BD CellQuest Pro software on Mac® OS 9, refer to the BD CellQuest Pro software (version 5.1 or later) ReadMe file for a description of new features. ReadMe files are located in the Documentation folder/Read Me Files folder on your hard drive. Read Me Files folder This tutorial covers: • Displaying data • Analyzing data • Additional features (enhanced Snap-To gating features, expression editor, load sample, and so on) To perform the steps in this tutorial, you will need BD CellQuest Pro software, version 5.1 or higher. BD CellQuest Pro software allows you to acquire and analyze data from your flow cytometer on a Macintosh® computer. In BD CellQuest Pro software, an Experiment document contains elements such as plots, regions, gates, statistics, markers, and annotated text. Data files are not saved in the Experiment document. The software saves a reference to the data files that contain the information needed to recreate the plots and stats on the Experiment document. Acquisition and analysis of flow cytometry data is a multistep process, as shown in Figure 1-1 on page 2. This tutorial focuses on analysis. BD CellQuest Pro Software Analysis Tutorial 336485 Rev. A 1 Start Up Perform Optimize Acquire Analyze Shut Down System QC Settings Data Data System • Display data. • Analyze data. – Create regions, gates, statistics views, user-defined expressions, and so on. – Set up a batch analysis, if needed. – Perform QC of analysis (for example, verify gates are set appropriately). -

Website and Ektron 9.1
WEBSITE AND EKTRON 9.1 TRAINING AND REFERENCE DOCUMENTATION Created by Hendrix College 1 Last updated September 18, 2014 TABLE OF CONTENTS Introduction ................................................................................................................................................................... 7 Your role in the Hendrix website ............................................................................................................................... 7 Orientation .................................................................................................................................................................... 8 Parts of a web page ................................................................................................................................................... 8 Page Content ......................................................................................................................................................... 8 Section Menu ......................................................................................................................................................... 8 Header and Footer ................................................................................................................................................. 8 Types of Content........................................................................................................................................................ 8 Content ................................................................................................................................................................. -

Why Delphi? Mobile Apps Are Everywhere
Expert Delphi Robust and fast cross-platform application development Paweł Głowacki BIRMINGHAM - MUMBAI Expert Delphi Copyright © 2017 Packt Publishing All rights reserved. No part of this book may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, without the prior written permission of the publisher, except in the case of brief quotations embedded in critical articles or reviews. Every effort has been made in the preparation of this book to ensure the accuracy of the information presented. However, the information contained in this book is sold without warranty, either express or implied. Neither the author, nor Packt Publishing, and its dealers and distributors will be held liable for any damages caused or alleged to be caused directly or indirectly by this book. Packt Publishing has endeavored to provide trademark information about all of the companies and products mentioned in this book by the appropriate use of capitals. However, Packt Publishing cannot guarantee the accuracy of this information. First published: June 2017 Production reference: 1300617 Published by Packt Publishing Ltd. Livery Place 35 Livery Street Birmingham B3 2PB, UK. ISBN 978-1-78646-016-5 www.packtpub.com Credits Author Copy Editors Paweł Głowacki Safis Editing Muktikant Garimella Reviewer Project Coordinator Dave Nottage Vaidehi Sawant Commissioning Editor Proofreader Kunal Parikh Safis Editing Acquisition Editor Indexer Nitin Dasan Aishwarya Gangawane Content Development Editor Graphics Anurag Ghogre Abhinash Sahu Technical Editors Production Coordinator Madhunikita Sunil Chindarkar Shantanu Zagade Rutuja Vaze Foreword I have known and worked with Paweł Głowacki for more than 16 years. Paweł is one of the world-wide Delphi community leading experts. -

Network Detective Inspector User Guide
USER GUIDE Inspector Deep-Dive Scans Produce Special Reports for Added Network & Security Expertise 9/20/2021 3:05 PM Network Detective Inspector — User Guide Contents Introduction to Inspector 5 Inspector Overview 5 Components of the Inspector Software Appliance 5 Inspector Software Appliance Features 7 Network Assessment Network Scan 7 Layer 2/3 Discovery of Network Devices (Exclusive to the Inspector) 7 Internal Vulnerability Scan (Exclusive to the Inspector) 7 HIPAA Compliance and Risk Assessment Scans 8 PCI Compliance and Risk Assessment Scans 8 External Vulnerability Scan 8 Automated Assessment Reporting 8 Remote Updating of the Inspector Software Appliance 9 Inspector Automated Scanning and Scheduling Best Practices 9 Inspector Appliance System Requirements 9 Setting Up Inspector 11 Initial Inspector Set Up 11 Step 1 — Install Inspector Appliance on MSP Network 11 Step 2 — Open Existing Network Detective Site with an Active Assessment Project 13 Step 3 — Associate Inspector with a Site 14 Configure Inspector Scans 15 Inspector Scans 19 Managing, Running, and Scheduling Scans (Inspector) 19 Scan Task Library versus Scan Tasks Queue 19 Manage the Scan Queue 19 Run a Scan On-Demand 20 Schedule a Scan 21 Cancel a Scan 23 Configuring Inspector Scans by Type/Assessment Module 25 2 Network/Security Scan 25 Scanning an Active Directory Domain Network 25 Scanning a Workgroup Network 34 Using the Run Now Option 42 HIPAA Compliance Network Scan 44 PCI Compliance Network Scan 46 Push Deploy Local Computer Scan for PCI 47 Push Deploy Local Computer -

Sportscodemanual-2.Pdf
Part 1 - An Introduction 1 Overview of SportsCode 2 SportsCode Set-up Requirements 3 Registration of SportsCode V10 4 SportsCode Preferences 5 General ������������������������������������������������������������������������������������������������������������������������������������������������������������������������������5 Capture ������������������������������������������������������������������������������������������������������������������������������������������������������������������������������6 Movies �������������������������������������������������������������������������������������������������������������������������������������������������������������������������������7 Timelines ���������������������������������������������������������������������������������������������������������������������������������������������������������������������������9 Overlay Text * ������������������������������������������������������������������������������������������������������������������������������������������������������������������9 Flagging * �����������������������������������������������������������������������������������������������������������������������������������������������������������������������11 Communications �������������������������������������������������������������������������������������������������������������������������������������������������������� 12 Share ������������������������������������������������������������������������������������������������������������������������������������������������������������������������������� -

Palm-Tech Inspector
Palm-Tech Inspector Portable Inspection Software User’s Guide Version 5.0 March 2007 Version 1.05 Copyrights and Trademarks Palm-Tech Inspector Software is a trademark of PDmB, Inc. Palm-Tech Inspector software is the property of PDmB, Inc. and is Copyright © 1998–2007. All rights reserved. Palm-Tech Picture Album software is the property of PDmB, Inc. and is Copyright © 1999–2007. All rights reserved. Palm-Tech Inspection Designer software is the property of PDmB, Inc. and is Copyright © 2002–2007. All rights reserved. Palm-Tech Front Office software is the property of PDmB, Inc. and is Copyright © 2002–2007. All rights reserved. Manual Copyright © 1998–2007 by PDmB, Inc. Palm-Tech Inspector software, manual, or portions thereof may not be reproduced in any form whatsoever (except as allowed for archive purposes) without the written permission of PDmB, Inc. Palm-Tech, Palm-Tech Inspector, Palm-Tech Inspection Designer, Palm-Tech Picture Album and Palm-Tech Front Office are trademarks of PDmB, Inc. Windows ™ is a registered trademark of Microsoft Corporation. Correspondence concerning Palm-Tech Inspector should be directed to: PDmB, Inc. 9600 Colerain Ave., Suite 110 Cincinnati, OH 45251 513-522-7362 e-mail [email protected] support inquiries go to [email protected] ii Palm-Tech Inspector software and manual are licensed property of PDmB, Inc. Use of the software indicates your acceptance of the following END USER LICENSE AGREEMENT. END USER LICENSE AGREEMENT CAREFULLY READ THE FOLLOWING LICENSE AGREEMENT. BY OPENING THE PACKAGE YOU ARE CONSENTING TO BE BOUND BY AND ARE BECOMING A PARTY TO THIS AGREEMENT. -

Appendix a Bacnet OPC Data Access Server
M-Explorer User’s Guide A-1 Appendix A BACnet OPC Data Access Server Introduction The BACnet OPC Data Server provides M-Explorer with dynamic data values from N30 series supervisory controller objects, third-party BACnet objects, and Cardkey PEGASYS objects for display. M-Inspector views and commands specific N30, BACnet, and Cardkey PEGASYS objects. In addition to describing how M-Explorer and M-Inspector provide unique access to the data provided by the BACnet OPC Data Access Server, this appendix describes how to: • command an object • view an object • edit an object © January, 2001 Johnson Controls, Inc. www.johnsoncontrols.com A-2 M-Explorer Key Concepts M-Explorer JC.BNOPC Hierarchy M-Explorer presents data from a BACnet OPC Data Access Server in a hierarchical structure as illustrated in Figure A-1. Table A-1 describes the numbered callouts in Figure A-1. This hierarchy matches the M-Tool Project Builder application and the VT100 interface embedded in the N30 supervisory controller. However, this hierarchy is different from the OPC Tag Browser which shows a flat list of named objects within each N30 device and does not show additional containers within the N30 device. Third-party BACnet devices and Cardkey devices are shown in a flat list similar to the Tag Browser. Refer to Project Builder User’s Guide (LIT-693200) in the M-Tool Manual (FAN 693), the N30 Supervisory Controller User’s Manual (FAN 689.2), and M-Graphics User’s Manual (FAN 644.0) for more information. Figure A-1: M-Explorer JC.BNOPC Hierarchy Appendix A BACnet OPC Data Access Server A-3 Table A-1: M-Explorer JC.BNOPC Hierarchy Callouts Callout View/Item Description 1 M-Explorer Top of the M-Explorer hierarchy 2 JC.BNOPC BACnet OPC Data Access server 3 Phoenix Devices Site (name) 4 GEX, HDMAV, etc. -

Switch User Guide 182925 February 2016
User Guide Switch 2.1 User Guide for Mac Switch User Guide 182925 February 2016 3 Contents Preface 7 Copyrights and Trademark Notices 7 Telestream Contact Information 11 Overview 13 Introduction 13 Play License Restrictions 14 Switch Plus Features 14 Switch Pro Features 15 System Requirements 17 Summary of License Features 18 Installing Switch 19 Introduction 19 Installing Switch 19 Uninstalling Switch 20 Updating/Activating/Deactivating Switch 21 Opening Switch 21 Opening Files 25 Introduction 25 Opening Files 25 Opening Secondary Files 25 Opening Recent Files 26 Closing Files 26 Media Playback 27 Introduction 27 Play Bar 27 4 Contents Control Menu 28 Timeline 31 Audio Control 32 Inspecting Media 33 Introduction 33 Media Inspector 33 Inspect/Edit Mode Switch 35 Container Tab 35 File Controls 35 Metadata and Secondary Audio/Subtitles 36 Video Tab 37 Audio Tab 38 Subtitle Tab 39 Time Tab 40 Editing Media 43 Introduction 43 Media Inspector 43 Inspect/Edit Mode Switch 45 Container Tab 45 File Controls 45 Metadata and Secondary Audio/Subtitles 46 Video Tab 47 Audio Tab 50 Subtitle Tab 52 Time Tab 54 Exporting Media 57 Introduction 57 Exporting Media 57 Exporting Movies 58 Exporting iTunes Packages 58 Exporting an iTunes Asset Only .itmsp Package 60 Exporting Video Frames 61 Getting Help 63 Introduction 63 Getting Help 64 Searching for Help 64 User Guides 64 Purchasing Switch 65 Customer Support 66 Switch User Guide Contents 5 Preferences and Updates 69 Introduction 69 Setting Preferences 69 Update 69 License 70 Activating a Serial Number -

A Practical Introduction to 3D Game Development
Yasser Jaffal A Practical Introduction to 3D Game Development 2 Download free eBooks at bookboon.com A Practical Introduction to 3D Game Development 1st edition © 2014 Yasser Jaffal & bookboon.com ISBN 978-87-403-0786-3 3 Download free eBooks at bookboon.com A Practical Introduction to 3D Game Development Contents Contents About this Book 7 1 Basics of Scene Construction 8 1.1 Basic shapes and their properties 8 1.2 Relations between game objects 11 1.3 Rendering properties 13 1.4 Light types and properties 16 1.5 Camera 19 1.6 Controlling objects properties 21 2 Handling User Input 28 2.1 Reading keyboard input 29 2.2 Implementing platformer input system 32 2.3 Reading mouse input 43 2.4 Implementing first person shooter input system 46 �e Graduate Programme I joined MITAS because for Engineers and Geoscientists I wanted real responsibili� www.discovermitas.comMaersk.com/Mitas �e Graduate Programme I joined MITAS because for Engineers and Geoscientists I wanted real responsibili� Maersk.com/Mitas Month 16 I wwasas a construction Month 16 supervisorI wwasas in a construction the North Sea supervisor in advising and the North Sea Real work helpinghe foremen advising and IInternationalnternationaal opportunities ��reeree wworkoro placements solves Real work problems helpinghe foremen IInternationalnternationaal opportunities ��reeree wworkoro placements solves problems 4 Click on the ad to read more Download free eBooks at bookboon.com A Practical Introduction to 3D Game Development Contents 2.5 Implementing third person input system 53