Alimera Sciences Inc
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

NASDAQ Stock Market
Nasdaq Stock Market Friday, December 28, 2018 Name Symbol Close 1st Constitution Bancorp FCCY 19.75 1st Source SRCE 40.25 2U TWOU 48.31 21st Century Fox Cl A FOXA 47.97 21st Century Fox Cl B FOX 47.62 21Vianet Group ADR VNET 8.63 51job ADR JOBS 61.7 111 ADR YI 6.05 360 Finance ADR QFIN 15.74 1347 Property Insurance Holdings PIH 4.05 1-800-FLOWERS.COM Cl A FLWS 11.92 AAON AAON 34.85 Abiomed ABMD 318.17 Acacia Communications ACIA 37.69 Acacia Research - Acacia ACTG 3 Technologies Acadia Healthcare ACHC 25.56 ACADIA Pharmaceuticals ACAD 15.65 Acceleron Pharma XLRN 44.13 Access National ANCX 21.31 Accuray ARAY 3.45 AcelRx Pharmaceuticals ACRX 2.34 Aceto ACET 0.82 Achaogen AKAO 1.31 Achillion Pharmaceuticals ACHN 1.48 AC Immune ACIU 9.78 ACI Worldwide ACIW 27.25 Aclaris Therapeutics ACRS 7.31 ACM Research Cl A ACMR 10.47 Acorda Therapeutics ACOR 14.98 Activision Blizzard ATVI 46.8 Adamas Pharmaceuticals ADMS 8.45 Adaptimmune Therapeutics ADR ADAP 5.15 Addus HomeCare ADUS 67.27 ADDvantage Technologies Group AEY 1.43 Adobe ADBE 223.13 Adtran ADTN 10.82 Aduro Biotech ADRO 2.65 Advanced Emissions Solutions ADES 10.07 Advanced Energy Industries AEIS 42.71 Advanced Micro Devices AMD 17.82 Advaxis ADXS 0.19 Adverum Biotechnologies ADVM 3.2 Aegion AEGN 16.24 Aeglea BioTherapeutics AGLE 7.67 Aemetis AMTX 0.57 Aerie Pharmaceuticals AERI 35.52 AeroVironment AVAV 67.57 Aevi Genomic Medicine GNMX 0.67 Affimed AFMD 3.11 Agile Therapeutics AGRX 0.61 Agilysys AGYS 14.59 Agios Pharmaceuticals AGIO 45.3 AGNC Investment AGNC 17.73 AgroFresh Solutions AGFS 3.85 -
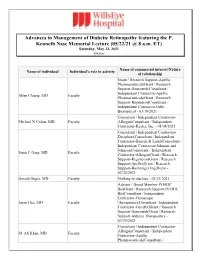
Advances in Management of Diabetic Retinopathy Featuring the P
Advances in Management of Diabetic Retinopathy featuring the P. Kenneth Nase Memorial Lecture (05/22/21 @ 8 a.m. ET) Saturday, May 22, 2021 Online Name of commercial interest/Nature Name of individual Individual's role in activity of relationship Grant / Research Support-Apellis Pharmaceuticals|Grant / Research Support-Genentech|Consultant / Independent Contractor-Apellis Allen Chiang, MD Faculty Pharmaceuticals|Grant / Research Support-Regeneron|Consultant / Independent Contractor-Orbit Biomedical - 03/19/2021 Consultant / Independent Contractor- Michael N Cohen, MD Faculty Allergan|Consultant / Independent Contractor-Keeler, Inc. - 05/04/2021 Consultant / Independent Contractor- Deciphera|Consultant / Independent Contractor-Bausch & Lomb|Consultant / Independent Contractor-Johnson and Johnson|Consultant / Independent Sunir J. Garg, MD Faculty Contractor-Allergan|Grant / Research Support-Regeneron|Grant / Research Support-Apellis|Grant / Research Support-Boehringer Ingelheim - 02/24/2021 Omesh Gupta, MD Faculty Nothing to disclose - 03/21/2021 Advisor / Board Member-IVERIC Bio|Grant / Research Support-IVERIC Bio|Consultant / Independent Contractor-Gyroscope Jason Hsu, MD Faculty Therapeutics|Consultant / Independent Contractor-OccuRx|Grant / Research Support-Genentech|Grant / Research Support-Aldeyra Therapeutics - 03/20/2021 Consultant / Independent Contractor- Allergan|Consultant / Independent M. Ali Khan, MD Faculty Contractor-Apellis Pharmaceuticals|Consultant / Independent Contractor- Genentech|Grant / Research Support- Regeneron -

Medicaid System (Mmis) Illinois Department of Run Date: 08/08/2015 Provider Subsystem Healthcare and Family Services Run Time: 21:25:58 Report Id 2794D052 Page: 01
MEDICAID SYSTEM (MMIS) ILLINOIS DEPARTMENT OF RUN DATE: 08/08/2015 PROVIDER SUBSYSTEM HEALTHCARE AND FAMILY SERVICES RUN TIME: 21:25:58 REPORT ID 2794D052 PAGE: 01 ALPHA COMPLETE LIST OF PHARMACEUTICAL LABELERS WITH SIGNED REBATE AGREEMENTS IN EFFECT AS OF 10/01/2015 NDC NDC PREFIX LABELER NAME PREFIX LABELER NAME 68782 (OSI) EYETECH 65162 AMNEAL PHARMACEUTICALS LLC 00074 ABBOTT LABORATORIES 69238 AMNEAL PHARMACEUTICALS, LLC 68817 ABRAXIS BIOSCIENCE, LLC 53150 AMNEAL-AGILA, LLC 16729 ACCORD HEALTHCARE INCORPORATED 00548 AMPHASTAR PHARMACEUTICALS, INC. 42192 ACELLA PHARMACEUTICALS, LLC 66780 AMYLIN PHARMACEUTICALS, INC. 10144 ACORDA THERAPEUTICS, INC. 55724 ANACOR PHARMACEUTICALS 00472 ACTAVIS 10370 ANCHEN PHARMACEUTICALS, INC. 00228 ACTAVIS ELIZABETH LLC 62559 ANIP ACQUISITION COMPANY 45963 ACTAVIS INC. 54436 ANTARES PHARMA, INC. 46987 ACTAVIS KADIAN LLC 52609 APO-PHARMA USA, INC. 49687 ACTAVIS KADIAN LLC 60505 APOTEX CORP. 14550 ACTAVIS PHARMA MFGING PRIVATE LIMITED 63323 APP PHARMACEUTICALS, LLC. 67767 ACTAVIS SOUTH ATLANTIC 42865 APTALIS PHARMA US, INC 66215 ACTELION PHARMACEUTICALS U.S., INC. 58914 APTALIS PHARMA US, INC. 52244 ACTIENT PHARMACEUTICALS 13310 AR SCIENTIFIC, INC. 75989 ACTON PHARMACEUTICALS 08221 ARBOR PHARM IRELAND LIMITED 76431 AEGERION PHARMACEUTICALS, INC. 60631 ARBOR PHARMACEUTICALS IRELAND LIMITED 50102 AFAXYS, INC. 24338 ARBOR PHARMACEUTICALS, INC. 10572 AFFORDABLE PHARMACEUTICALS, LLC 59923 AREVA PHARMACEUTICALS 27241 AJANTA PHARMA LIMITED 76189 ARIAD PHARMACEUTICALS, INC. 17478 AKORN INC 24486 ARISTOS PHARMACEUTICALS, INC. 24090 AKRIMAX PHARMACEUTICALS LLC 67877 ASCEND LABORATORIES, L.L.C. 68220 ALAVEN PHARMACEUTICAL, LLC 76388 ASPEN GLOBAL INC. 00065 ALCON LABORATORIES, INC. 51248 ASTELLAS 00998 ALCON LABORATORIES, INC. 00469 ASTELLAS PHARMA US, INC. 25682 ALEXION PHARMACEUTICALS 00186 ASTRAZENECA LP 68611 ALIMERA SCIENCES, INC. -

05/09/2016 Provider Subsystem Healthcare and Family Services Run Time: 04:25:50 Report Id 2794D052 Page: 01
MEDICAID SYSTEM (MMIS) ILLINOIS DEPARTMENT OF RUN DATE: 05/09/2016 PROVIDER SUBSYSTEM HEALTHCARE AND FAMILY SERVICES RUN TIME: 04:25:50 REPORT ID 2794D052 PAGE: 01 ALPHA COMPLETE LIST OF PHARMACEUTICAL LABELERS WITH SIGNED REBATE AGREEMENTS IN EFFECT AS OF 07/01/2016 NDC NDC PREFIX LABELER NAME PREFIX LABELER NAME 68782 (OSI) EYETECH 55513 AMGEN USA 00074 ABBOTT LABORATORIES 58406 AMGEN/IMMUNEX 68817 ABRAXIS BIOSCIENCE, LLC 53746 AMNEAL PHARMACEUTICALS 16729 ACCORD HEALTHCARE INCORPORATED 65162 AMNEAL PHARMACEUTICALS LLC 42192 ACELLA PHARMACEUTICALS, LLC 69238 AMNEAL PHARMACEUTICALS, LLC 10144 ACORDA THERAPEUTICS, INC. 53150 AMNEAL-AGILA, LLC 00472 ACTAVIS 00548 AMPHASTAR PHARMACEUTICALS, INC. 00228 ACTAVIS ELIZABETH LLC 69918 AMRING PHARMACEUTICALS, INC. 45963 ACTAVIS INC. 66780 AMYLIN PHARMACEUTICALS, INC. 46987 ACTAVIS KADIAN LLC 55724 ANACOR PHARMACEUTICALS 49687 ACTAVIS KADIAN LLC 10370 ANCHEN PHARMACEUTICALS, INC. 14550 ACTAVIS PHARMA MFGING PRIVATE LIMITED 43595 ANGELINI PHARMA, INC. 61874 ACTAVIS PHARMA, INC. 62559 ANIP ACQUISITION COMPANY 67767 ACTAVIS SOUTH ATLANTIC 54436 ANTARES PHARMA, INC. 66215 ACTELION PHARMACEUTICALS U.S., INC. 52609 APO-PHARMA USA, INC. 52244 ACTIENT PHARMACEUTICALS 60505 APOTEX CORP. 75989 ACTON PHARMACEUTICALS 63323 APP PHARMACEUTICALS, LLC. 69547 ADAPT PHARMA INC. 43485 APRECIA PHARMACEUTICALS COMPANY 76431 AEGERION PHARMACEUTICALS, INC. 42865 APTALIS PHARMA US, INC 50102 AFAXYS, INC. 58914 APTALIS PHARMA US, INC. 10572 AFFORDABLE PHARMACEUTICALS, LLC 13310 AR SCIENTIFIC, INC. 27241 AJANTA PHARMA LIMITED 08221 ARBOR PHARM IRELAND LIMITED 17478 AKORN INC 60631 ARBOR PHARMACEUTICALS IRELAND LIMITED 24090 AKRIMAX PHARMACEUTICALS LLC 24338 ARBOR PHARMACEUTICALS, INC. 68220 ALAVEN PHARMACEUTICAL, LLC 59923 AREVA PHARMACEUTICALS 00065 ALCON LABORATORIES, INC. 76189 ARIAD PHARMACEUTICALS, INC. 00998 ALCON LABORATORIES, INC. 24486 ARISTOS PHARMACEUTICALS, INC. -

Rebateable Manufacturers
Rebateable Labelers – July 2021 Manufacturers are responsible for updating their eligible drugs and pricing with CMS. Montana Healthcare Programs will not pay for an NDC not updated with CMS. Note: Some manufacturers on this list may have some NDCs that are covered and others that are not. Manufacturer ID Manufacturer Name 00002 ELI LILLY AND COMPANY 00003 E.R. SQUIBB & SONS, LLC. 00004 HOFFMANN-LA ROCHE 00006 MERCK & CO., INC. 00007 GLAXOSMITHKLINE 00008 WYETH PHARMACEUTICALS LLC, 00009 PHARMACIA AND UPJOHN COMPANY LLC 00013 PFIZER LABORATORIES DIV PFIZER INC 00015 MEAD JOHNSON AND COMPANY 00023 ALLERGAN INC 00024 SANOFI-AVENTIS, US LLC 00025 PFIZER LABORATORIES DIV PFIZER INC 00026 BAYER HEALTHCARE LLC 00032 ABBVIE INC. 00037 MEDA PHARMACEUTICALS, INC. 00039 SANOFI-AVENTIS, US LLC 00046 WYETH PHARMACEUTICALS INC. 00049 ROERIG 00051 ABBVIE INC 00052 ORGANON USA INC. 00053 CSL BEHRING L.L.C. 00054 HIKMA PHARMACEUTICAL USA, INC. 00056 BRISTOL-MYERS SQUIBB PHARMA CO. 00065 ALCON LABORATORIES, INC. 00068 AVENTIS PHARMACEUTICALS 00069 PFIZER LABORATORIES DIV PFIZER INC 00071 PARKE-DAVIS DIV OF PFIZER 00074 ABBVIE INC 00075 AVENTIS PHARMACEUTICALS, INC. 00078 NOVARTIS 00085 SCHERING CORPORATION 00087 BRISTOL-MYERS SQUIBB COMPANY 00088 AVENTIS PHARMACEUTICALS 00093 TEVA PHARMACEUTICALS USA, INC. 00095 BAUSCH HEALTH US, LLC Page 1 of 19 Manufacturer ID Manufacturer Name 00096 PERSON & COVEY, INC. 00113 L. PERRIGO COMPANY 00115 IMPAX GENERICS 00116 XTTRIUM LABORATORIES, INC. 00121 PHARMACEUTICAL ASSOCIATES, INC. 00131 UCB, INC. 00132 C B FLEET COMPANY INC 00143 HIKMA PHARMACEUTICAL USA, INC. 00145 STIEFEL LABORATORIES, INC, 00168 E FOUGERA AND CO. 00169 NOVO NORDISK, INC. 00172 TEVA PHARMACEUTICALS USA, INC 00173 GLAXOSMITHKLINE 00178 MISSION PHARMACAL COMPANY 00185 EON LABS, INC. -

Psivida and Pfizer Amend Agreement to Focus Solely on Development Of
June 14, 2011 pSivida and Pfizer Amend Agreement to Focus Solely on Development of Sustained- Release Implant to Deliver Latanoprost for Ocular Hypertension and Glaucoma pSivida to receive up to $168m plus royalties WATERTOWN, Mass., Jun 14, 2011 (BUSINESS WIRE) -- Drug delivery company pSivida Corp (NASDAQ:PSDV)(ASX:PVA) today announced it amended and restated its Research and Development Agreement with Pfizer Inc. (NYSE:PFE) to focus solely on the development of a long-term, sustained-release implant to deliver latanoprost for patients with ocular hypertension and glaucoma. The proposed implant is a bioerodible version of pSivida's proprietary Durasert™ technology system and is designed to be injected into the subconjunctival space of the eye. Under this revised agreement, Pfizer will make an initial payment of $2.3 million. pSivida will, with technical assistance from Pfizer, have the right to develop the glaucoma product candidate through Phase II clinical trials. At that point, Pfizer may exercise its option for an exclusive, worldwide license to develop and commercialize the product candidate in return for a $20 million payment, double-digit royalty payments on any sales of the product and additional development, regulatory and sales performance milestone payments of up to $146.5 million. If Pfizer does not exercise its option, pSivida will retain the right to develop and commercialize the glaucoma product on its own or with a partner. As part of the amended agreement, pSivida regains all rights to its intellectual property in ophthalmic applications previously included in the original Research and Collaboration Agreement other than that required for the latanoprost implant. -

The Bottom 99
The Top 100 November, 2013 A list of stocks topping our custom 'torpedo’ screen. Updated monthly. BAGR Diversified Restaurant Holdings, Inc. Consumer Discretionary GRPN Groupon, Inc. Class A Consumer Discretionary JCP J. C. Penney Company, Inc. Consumer Discretionary SQBG Sequential Brands Group, Inc. Consumer Discretionary TPX Tempur Sealy International Inc Consumer Discretionary TTS Tile Shop Holdings, Inc. Consumer Discretionary ZQK Quiksilver, Inc. Consumer Discretionary APL Atlas Pipeline Partners, L.P. Energy ARP Atlas Resource Partners, L.P. Energy ATLS Atlas Energy, L.P. Energy CMLP Crestwood Midstream Partners LP Energy CQP Cheniere Energy Partners, L.P. Energy GDP Goodrich Petroleum Corporation Energy GPOR Gulfport Energy Corporation Energy KIOR KiOR, Inc. Class A Energy LNG Cheniere Energy, Inc. Energy MWE MarkWest Energy Partners, L.P. Energy PVR PVR Partners, L.P. Energy SZYM Solazyme, Inc. Energy AMT American Tower Corporation Financials Z Zillow, Inc. Class A Financials ACAD ACADIA Pharmaceuticals Inc. Health Care ACHN Achillion Pharmaceuticals, Inc. Health Care AGEN Agenus Inc. Health Care ALIM Alimera Sciences, Inc. Health Care ALNY Alnylam Pharmaceuticals, Inc Health Care ARAY Accuray Incorporated Health Care ARIA ARIAD Pharmaceuticals, Inc. Health Care ATHN athenahealth, Inc. Health Care ATHX Athersys, Inc. Health Care ATRS Antares Pharma, Inc. Health Care AXGN AxoGen, Inc. Health Care BAXS Baxano Surgical, Inc. Health Care BDSI BioDelivery Sciences International, Inc. Health Care BIOS BioScrip, Inc. Health Care BMRN BioMarin Pharmaceutical Inc. Health Care CCXI Chemocentryx, Inc. Health Care CRIS Curis, Inc. Health Care CSU Capital Senior Living Corporation Health Care CUR Neuralstem, Inc. Health Care CYCC Cyclacel Pharmaceuticals, Inc. Health Care DSCI Derma Sciences, Inc. -

MANUFACTURERS with CMS AGREEMENTS AS of 5-14-21 Labeler Code Labeler Name Program End Date 00002 ELI LILLY and COMPANY 00003 E.R
MANUFACTURERS WITH CMS AGREEMENTS AS OF 5-14-21 Labeler Code Labeler Name Program End Date 00002 ELI LILLY AND COMPANY 00003 E.R. SQUIBB & SONS, LLC. 00004 HOFFMANN-LA ROCHE 00006 MERCK & CO., INC. 00007 GLAXOSMITHKLINE 00008 WYETH PHARMACEUTICALS LLC, 00009 PHARMACIA AND UPJOHN COMPANY LLC 00013 PFIZER LABORATORIES DIV PFIZER INC 00015 MEAD JOHNSON AND COMPANY 00023 ALLERGAN INC 00024 SANOFI-AVENTIS, US LLC 00025 PFIZER LABORATORIES DIV PFIZER INC 00026 BAYER HEALTHCARE LLC 00032 ABBVIE INC. 00037 MEDA PHARMACEUTICALS, INC. 00039 SANOFI-AVENTIS, US LLC 00046 WYETH PHARMACEUTICALS INC. 00049 ROERIG 00051 ABBVIE INC 00052 ORGANON USA INC. 00053 CSL BEHRING L.L.C. 00054 HIKMA PHARMACEUTICAL USA, INC. 00056 BRISTOL-MYERS SQUIBB PHARMA CO. 00065 ALCON LABORATORIES, INC. 00068 AVENTIS PHARMACEUTICALS 00069 PFIZER LABORATORIES DIV PFIZER INC 00071 PARKE-DAVIS DIV OF PFIZER 00074 ABBVIE INC 00075 AVENTIS PHARMACEUTICALS, INC. 00078 NOVARTIS 00085 SCHERING CORPORATION 00087 BRISTOL-MYERS SQUIBB COMPANY 00088 AVENTIS PHARMACEUTICALS 00093 TEVA PHARMACEUTICALS USA, INC. 00095 BAUSCH HEALTH US, LLC 00096 PERSON & COVEY, INC. 00113 L. PERRIGO COMPANY 00115 IMPAX GENERICS 00116 XTTRIUM LABORATORIES, INC. 00121 PHARMACEUTICAL ASSOCIATES, INC. 00131 UCB, INC. 00132 C B FLEET COMPANY INC 00143 HIKMA PHARMACEUTICAL USA, INC. 00145 STIEFEL LABORATORIES, INC, 00168 E FOUGERA AND CO. 00169 NOVO NORDISK, INC. 00172 TEVA PHARMACEUTICALS USA, INC Labeler Code Labeler Name Program End Date 00173 GLAXOSMITHKLINE 00178 MISSION PHARMACAL COMPANY 00185 EON LABS, INC. 00186 ASTRAZENECA PHARMACEUTICALS LP 00187 BAUSCH HEALTH US, LLC. 00206 WYETH PHARMACEUTICALS LLC, 00224 KONSYL PHARMACEUTICALS, INC. 00225 B. F. ASCHER AND COMPANY, INC. 00228 ACTAVIS PHARMA, INC. -

Table of Contents
Table of contents Executive Summary Chapter 1 – Introduction Chapter 2 – Trends in drug delivery dealmaking 2.1. Introduction 2.2. Drug delivery partnering over the years 2.3. Bigpharma drug delivery dealmaking activity 2.4. Big biotech drug delivery dealmaking activity 2.5. Most active drug delivery dealmakers 2.6 Drug delivery partnering by deal type 2.7. Drug delivery partnering by disease type 2.8. Partnering by drug delivery technology type 2.9. Average deal terms for drug delivery partnering 2.9.1 Drug delivery headline values 2.9.2 Drug delivery upfront payments 2.9.3 Drug delivery milestone payments 2.9.4 Drug delivery royalty rates 2.10. The anatomy of drug delivery partnering 2.11. Drug delivery or specialty pharma? 2.11.1. Is specialty pharma the only way for drug delivery? 2.11.2. Best practice for optimizing drug delivery program development 2.11.3. The anatomy of a drug delivery deal 2.11.3.a. Case study 1: Alpharma – Durect 2.11.3.b. Case study 2: Bayer – MDRNA 2.11.3.c. Case study 3: Endo Pharmaceuticals – BioDelivery Sciences Chapter 3 – Leading drug delivery deals 3.1. Introduction 3.2. Top drug delivery deals by value Chapter 4 – Bigpharma drug delivery deals 4.1. Introduction 4.2. How to use bigpharma drug delivery partnering deals 4.3. Big pharma drug delivery partnering company profiles Abbott Actavis Inc (formerly Watson Pharmaceuticals) Actavis (merged with Watson Pharmaceuticals Oct 2012) Actelion Allergan Amgen Astellas AstraZeneca Baxter International Bayer Biogen Idec Boehringer Ingelheim Bristol-Myers Squibb Celgene CSL Daiichi Sankyo Dainippon Sumitomo Eisai Eli Lilly Endo Pharmaceuticals Forest Laboratories Galderma Gilead Sciences GlaxoSmithKline Grifols Hospira Johnson & Johnson Kyowa Hakko Kirin Lundbeck Menarini Merck & Co Merck KGaA Mylan Novartis Novo Nordisk Otsuka Pfizer Purdue Roche Sanofi Shionogi Shire Takeda Teva UCB Valeant Warner Chilcott Chapter 5 – Bigbiotech drug delivery deals 5.1. -

Supplier Name
Supplier Name 3M SECURITY PRINTING AND SYSTEMS LIMITED AAH HOSPITAL SERVICE ABBOTT DIAGNOSTICS DIVISION ABBOTT LABORATORIES ABBVIE LIMITED ABOUT FACE SOLUTIONS LTD ACCORD HEALTHCARE LIMITED ACLARDIAN LTD ACTAVIS UK LTD ADDENBROOKE'S HOSPITAL AIR LIQUIDE AIR LIQUIDE HEALTHCARE (HOME) AIR PRODUCTS PLC ALCON ALCURA UK LIMITED (HOME) ALIMERA SCIENCES ALIUM MEDICAL ALLERGAN LIMITED ALLIANCE HEALTHCARE LTD ALLIANCE PHARMACEUTICALS LTD ALLOGA UK LTD (ALK ABELLO) ALLOGA UK LTD (UNIDRUG) AMGEN LTD ANIMAS (HOME) AQUILANT DISTRIBUTION ASEPTIC MEDICAL DEVICES AVENTIS PHARMA B BRAUN MEDICAL LTD BASSAIRE LIMITED BAUSCH AND LOMB UK LTD BAXA UK LIMITED BAXTER HEALTHCARE LTD BAYER PLC BAYER SCHERING PHARMA BEDFORD HOSPITAL NHS TRUST BIO DIAGNOSTICS BIO PRODUCTS LABORATORY BIOMONDE BIRMINGHAM CHILDRENS HOSPITAL NHS FOUNDATION TRUST BOEHRINGER INGELHEIM LTD BRACCO UK LTD BRISTOL MYERS SQUIBB PHARMACEUTICALS LTD BRITANNIA PHARMACEUTICALS LTD BRITISH OXYGEN CO.LTD BRITISH OXYGEN CO.LTD SPECIAL GASES BTG INTERNATIONAL LTD CALEA UK LIMITED CARDIFF UNIVERSITY CARDINAL HEALTH MARTINDALE PRODUCTS CELGENE LIMITED CHAPTER LIMITED CHIESI LIMITED CHURCHILL HOSPITAL CLARITY PHARMA LTD CLINIGEN GROUP PLC CLINIGEN HEALTHCARE LIMITED CLINIGEN HEALTHCARE LTD CSL BEHRING DEXCOM UK (HOME) DRUGSRUS LIMITED DURBIN PLC ECOLAB ELIS CLEANROOM FARNHAM REPACKAGING UNIT FERRING PHARMACEUTICALS LTD FORUM HEALTH PRODUCTS LIMITED FRESENIUS KABI LTD FRESENIUS MEDICAL CARE (UK) LTD (HOME) GB UK HEALTHCARE GE HEALTHCARE LTD GENMED.ME LTD GENZYME THERAPEUTICS GILEAD SCIENCES LTD GLASGOW -

Psivida Generates up to A$82M from Licensing Amendment with Alimera
March 17, 2008 pSivida generates up to A$82m from licensing amendment with Alimera ATLANTA / BOSTON, March 17, 2008 - Alimera Sciences and pSivida Ltd (NASDAQ:PSDV, ASX:PSD, Xetra:PSI) today announced that they have amended their license and collaboration agreement relating to Medidur™ FA, the companies’ Phase III investigative treatment for diabetic macular edema (DME), and other Medidur products. Alimera is increasing its equity in the future profits of Medidur FA from 50 to 80 percent in exchange for consideration of up to approximately A$82m (US$78m) to pSivida. Consideration to pSivida includes an up-front payment of A$13m (US$12m), a A$26m (US$25m) milestone payment upon FDA approval of Medidur™ FA, other payments of up to approximately A$22m (US$21m) by September 30, 2012, and assumption of pSivida's research and development funding obligations estimated at approximately A$21m (US$20m). “We are very pleased with this agreement as it provides us with the opportunity to increase our stake and consolidate the development and commercialization of our late stage DME product Medidur FA,”said Dan Myers, President and CEO of Alimera Sciences. “In addition, we will further advance the delivery system’s application in other serious ophthalmic conditions like dry age-related macular degeneration (AMD), using exploratory treatments such as the groundbreaking work we are doing around NADPH oxidase inhibitors.” "We believe this is a great deal for pSivida and its shareholders as it gives the company a significant financial interest in very exciting products and economics that eliminate our need for equity financing for the foreseeable future under our current plans,”said pSivida Managing Director, Dr. -

Fueling Life Sciences Through Transformative Transactions
Fueling life sciences through transformative transactions Q3 2016: TRENDS IN BIOTECH TRANSACTIONAL ACTIVITY October 25th, 2016 BOSTON l SAN FRANCISCO l TOKYO 1 Locust Walk focuses on building companies across three distinct segments Emerging Biopharma Growth Biopharma Medical Device Finance innovation by parallel Rapidly expand product mix Commercialize products processing global and and grow pipeline via strategic partnerships regional deals with private to maximize value and innovative capital and public capital tracks and mitigate risk sourcing to maximize value 2 Locust Walk is uniquely positioned to bring your transaction from search to signature Investment Banks Full-Time Hire • Significant financial • Industry operating analytic capability experience • Board level network • Deep company and contacts understanding through longer ‘engagement’ timeline Locust Walk Investment bank support, Board level strategy and insight, CBO expertise, flexible engagement term Consulting Firms Individual Consultant • Strategic analytics • Industry expertise and insights operating experience • Primary & secondary • Temporary research capabilities commitment • Board-ready deliverables 3 Locust Walk has helped build many successful biopharma companies Collaboration to develop & Advised on IPO process and Sell-side Asian Licensing Sell-side Asian Licensing Buy-side licensing agreement Buy-side product acquisition commercialize KHK’s anti- syndicate selection Agreement for fasinumab Agreement for Tecarfarin for ALT1103 for Acromegaly of Somatoprim for