Differential Regulation of Anthocyanins in Cerasus Humilis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
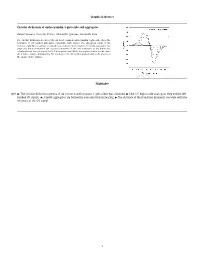
Graphical Abstract Circular Dichroism of Anthocyanidin 3-Glucoside Self
Graphical abstract Circular dichroism of anthocyanidin 3-glucoside self-aggregates Raquel Gavara, Vesselin Petrov, Alexandre Quintas, Fernando Pina * The circular dichroism spectra of the six most common anthocyanidin 3-glucoside show the formation of left handed aggregates compatible with dimers. The absorption bands of the monomer split by increasing concentration according to the formation of H and J aggregates. The angle and distance between the transition moments of the two monomers in the dimer was calculated from the splitting of the 0–0 absorption band. While the angle is similar for the series the distance changes dramatically. The intensity of the CD signal is proportional to the inverse of the square of the distance. Highlights Q10 " The circular dichroism spectra of six common anthocyanins 3-glucosides was obtained. " Like 3,5-diglucoside analogous they exhibit left- handed CD signals. " J and H aggregates are formed by concentration increasing. " The distance of the transition moments correlate with the intensity of the CD signal. 1 1 2 Circular dichroism of anthocyanidin 3-glucoside self-aggregates a a b a,⇑ 3 Q1 Raquel Gavara , Vesselin Petrov , Alexandre Quintas , Fernando Pina 4 a REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 2829516 Monte de Caparica, Portugal 5 b Instituto Superior de Ciencias da Saude Egas Moniz, Centro de Investigação Interdisciplinar Egas Moniz, P-2829511 Monte de Caparica, Caparica, Portugal 6 7 article info a b s t r a c t 1 9 10 Self-association constants for the flavylium cations of the six most common anthocyanidin 3-glucosides 20 11 were determined by circular dichroism (CD) and UV–Vis spectroscopy. -

Electrochemical and Antioxidant Properties of Anthocyanins and Anthocyanidins
CROATICA CHEMICA ACTA CCACAA 80 (1) 29¿34 (2007) ISSN-0011-1643 CCA-3135 Original Scientific Paper Electrochemical and Antioxidant Properties of Anthocyanins and Anthocyanidins Andréia A. de Lima, Eliana M. Sussuchi, and Wagner F. De Giovani* Departamento de Química, Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto, Universidade de São Paulo, Av. Bandeirantes 3900, 14040-901, Ribeirão Preto – SP, Brazil RECEIVED JUNE 26, 2006; REVISED SEPTEMBER 29, 2006; ACCEPTED OCTOBER 27, 2006 Electrochemical properties of delphinidin, cyanidin, pelargonidin, kuromanin and callistephin were investigated by cyclic and differential pulse voltammetries at different pH values and also in methanol. On the basis of oxidation potentials, the order of antioxidant activity for antho- cyanidins is delphinidin > cyanidin > pelargonidin. Oxidation peaks for anthocyanins (kuro- manin and callistephin) are shifted to more positive potentials compared to anthocyanidins (delphinidin, cyanidin and pelargonidin). Oxidation peak currents are linearly dependent on the Keywords square root of the scan rate, which is typical of a diffusion controlled electrochemical process. anthocyanins Antioxidant activities of the compounds were evaluated using the 1,1-diphenyl-2-picrylhydra- anthocyanidins zyl (DPPH) radical-scavenging method and they are directly related to their redox potential antioxidants values. The order of the antioxidant activity is delphinidin > cyanidin > pelargonidin > kuro- electrochemical properties manin > callistephin. INTRODUCTION pharmacological properties that render them interesting potential cancer chemopreventive agents.9–11 Because Anthocyanins are one of the main classes of flavonoids anthocyanins are widely consumed, finding out addi- in red wines. They have been pointed out as the molecu- tional biological activities related to these compounds lar entities that are most likely responsible for what has would be of great interest. -

Health-Promoting Effects of Traditional Foods
Health-PromotingFoodsTraditional Effects of • Marcello Iriti Health-Promoting Effects of Traditional Foods Edited by Marcello Iriti Printed Edition of the Special Issue Published in Foods Health-Promoting Effects of Traditional Foods Health-Promoting Effects of Traditional Foods Editor Marcello Iriti MDPI • Basel • Beijing • Wuhan • Barcelona • Belgrade • Manchester • Tokyo • Cluj • Tianjin Editor Marcello Iriti Department of Agricultural and Environmental Sciences, Milan State University Italy Editorial Office MDPI St. Alban-Anlage 66 4052 Basel, Switzerland This is a reprint of articles from the Special Issue published online in the open access journal Foods (ISSN 2304-8158) (available at: https://www.mdpi.com/journal/foods/special issues/health effects traditional foods). For citation purposes, cite each article independently as indicated on the article page online and as indicated below: LastName, A.A.; LastName, B.B.; LastName, C.C. Article Title. Journal Name Year, Article Number, Page Range. ISBN 978-3-03943-312-4 (Hbk) ISBN 978-3-03943-313-1 (PDF) c 2020 by the authors. Articles in this book are Open Access and distributed under the Creative Commons Attribution (CC BY) license, which allows users to download, copy and build upon published articles, as long as the author and publisher are properly credited, which ensures maximum dissemination and a wider impact of our publications. The book as a whole is distributed by MDPI under the terms and conditions of the Creative Commons license CC BY-NC-ND. Contents About the Editor .............................................. vii Marcello Iriti, Elena Maria Varoni and Sara Vitalini Healthy Diets and Modifiable Risk Factors for Non- Communicable Diseases—The European Perspective Reprinted from: Foods 2020, 9, 940, doi:10.3390/foods9070940 .................... -

Overexpression of Thmyc4e Enhances Anthocyanin Biosynthesis in Common Wheat
International Journal of Molecular Sciences Article Overexpression of ThMYC4E Enhances Anthocyanin Biosynthesis in Common Wheat 1,2,3, 1,2,3, 1,2,3 1,2 1,2,3 1,2,4, Shuo Zhao y, Xingyuan Xi y, Yuan Zong , Shiming Li , Yun Li , Dong Cao * and Baolong Liu 1,2,3,4,* 1 Qinghai Province Key Laboratory of Crop Molecular Breeding, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining 810008, China; [email protected] (S.Z.); [email protected] (X.X.); [email protected] (Y.Z.); [email protected] (S.L.); [email protected] (Y.L.) 2 Key Laboratory of Adaptation and Evolution of Plateau Biota, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining 810008, China 3 College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China 4 The Innovative Academy of Seed Design, Chinese Academy of Sciences, Xining 810008, China * Correspondence: [email protected] (D.C.); [email protected] (B.L.) These authors contributed equally to this work. y Received: 28 November 2019; Accepted: 23 December 2019; Published: 24 December 2019 Abstract: The basic helix-loop helix (bHLH) transcription factor has been inferred to play an important role in blue and purple grain traits in common wheat, but to date, its overexpression has not been reported. In this study, the bHLH transcription factor ThMYC4E, the candidate gene controlling the blue grain trait from Th. Ponticum, was transferred to the common wheat JW1. The positive transgenic lines displayed higher levels of purple anthocyanin pigments in their grains, leaves and glumes. -

Nutraceutical Components, Antioxidant Activity, and Color of 11 Varieties of Prickly Pear (Opuntia Sp.) 1 3 1* 2 M
Journal of Applied Botany and Food Quality 91, 211 - 218 (2018), DOI:10.5073/JABFQ.2018.091.028 1*Instituto de Horticultura, Universidad Autónoma Chapingo, Carretera México-Texcoco, Chapingo, Estado de México, México 2Instituto de Alimentos, Universidad Autónoma Chapingo, Carretera México-Texcoco, Chapingo, Estado de México, México 3Tecnológico Nacional de México, Instituto Tecnológico de Conkal, Avenida Tecnológico s/n Conkal, Yucatán, México Nutraceutical components, antioxidant activity, and color of 11 varieties of prickly pear (Opuntia sp.) 1 3 1* 2 M. Ramírez-Ramos , K. Medina-Dzul , R. García-Mateos , J. Corrales-García , C. Ybarra-Moncada2, A.M. Castillo-González1 (Submitted: October 13, 2017; Accepted: June 26, 2018) Summary tive diseases, due to the presence of certain metabolites (phenolic compounds, flavonoids, ascorbic acid, and betalains) (SUMAYA- In Mexico, there are 50 recorded varieties of the prickly pear fruit. MARTÍNEZ et al., 2011; ABOU-ELELLA and ALI, 2014). Biosynthetically, National production covers only the white-pulp fruit, but other betalamic acid-derived betalains, which are pigments used in the red varieties have export potential; however, their nutraceutical agri-food industry as natural colorants (STRACK et al., 2003), also properties are unknown. The pulp and peel (underutilized tissue) have antioxidant properties (LIVREA and TESORIERE, 2006; ALBANO of the pigmented fruits of the genus Opuntia sp. are marketed on a et al., 2015). Moreover, color is part of fruit quality and may therefore limited basis. They represent an alternative source of stable pigments be a factor in its preference, acceptance, or rejection, and play a (betalains), which are associated with antioxidant properties, for decisive role in the failure or success of prickly pear marketing. -

Characterization and Biological Activity of Condensed Tannins from Tropical Forage Legumes
1070 T.P. Pereira et al. Characterization and biological activity of condensed tannins from tropical forage legumes Tatiana Pires Pereira(1), Elisa Cristina Modesto(1), Delci de Deus Nepomuceno(2), Osniel Faria de Oliveira(3), Rafaela Scalise Xavier de Freitas(1), James Pierre Muir(4), José Carlos Batista Dubeux Junior(5) and João Carlos de Carvalho Almeida(1) (1)Universidade Federal Rural do Rio de Janeiro, Rodovia BR-465, Km 07, s/no, Zona Rural, CEP 23890-000 Seropédica, RJ, Brazil. E-mail: [email protected], [email protected], [email protected], [email protected] (2)In memoriam (3)Universidade Federal Rural de Pernambuco, Dois Irmãos, CEP 52171-900 Recife, PE, Brazil. E-mail: [email protected] (4)Texas AgriLife Research and Extension Center, Stephenville, TX, USA. E-mail: [email protected] (5)University of Florida, North Florida Research and Education Center, Highway, Marianna, FL, USA. E-mail: [email protected] Abstract – The objective of this work was to characterize condensed tannins (CT) from six tropical forage legumes and to determine their biological activity. The monomers propelargonidin, prodelphinidin and procyanidin were analyzed, as well as extractable condensed tannin (ECT), protein-bound CT (PBCT) and fiber-bound CT (FBCT), molecular weight, degree of polymerization, polydispersity index, and biological activity by protein precipitate by phenols (PPP) of leaves of the legumes Cajanus cajan, Gliricidia sepium, Stylosanthes capitata x Stylosanthes macrocephala (stylo), Flemingia macrophylla, Cratylia argentea, and Mimosa caesalpiniifolia, and of the bark of this latter species. Differences were observed in the concentrations of ECT, PBCT, PPP, and total condensed tannin among species, but not in that of FBCT. -

Callistephin Enhances the Protective Effects of Isoflurane on Microglial Injury Through Downregulation of Inflammation and Apoptosis
802 MOLECULAR MEDICINE REPORTS 20: 802-812, 2019 Callistephin enhances the protective effects of isoflurane on microglial injury through downregulation of inflammation and apoptosis LILI ZHAO, SHIBIAO CHEN, TIANYIN LIU, XIUHONG WANG, HAIJIN HUANG and WEICHENG LIU Department of Anesthesiology, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi 330006, P.R. China Received June 18, 2018; Accepted March 15, 2019 DOI: 10.3892/mmr.2019.10282 Abstract. Microglia are the major immune cells in the central enhanced the effects of isoflurane. Callistephin may therefore nervous system. Microglial activation can be beneficial or constitute a candidate drug agent that may target inflammatory detrimental depending on the stimuli and the physiopathological and growth regulatory signaling pathways, thus ameliorating environment. Microglial activation is involved in a variety certain aspects of neurodegenerative diseases. of neurodegenerative disorders. Different anesthetic agents have exhibited diverse effects on microglial activation and Introduction the engulfment process. The anthocyanin callistephin has been demonstrated to have antioxidant and anti‑inflammatory Microglial cells are the major immune cell in the central properties, and these were assessed in the present study, with a nervous system (CNS), responding against types of endog- focus on its effect on microglial activation. Mouse microglial enous and exogenous stimuli, including infection by bacteria, cells C8-4B were treated with 100 ng/µl lipopolysaccharide viruses, prions and β-amyloid plaques (1). Microglia are (LPS) and 1 ng/µl interferon-γ. Cells were subsequently treated activated upon exposure to different stimuli and, depending with 2% isoflurane, 100 µM callistephin or both. LPS promoted on the environmental context, this may be beneficial or detri- apoptosis in C8-B4 cells, and this was reduced following mental to the functionality and physiology of the CNS (2). -

Intereferents in Condensed Tannins Quantification by the Vanillin Assay
INTEREFERENTS IN CONDENSED TANNINS QUANTIFICATION BY THE VANILLIN ASSAY IOANNA MAVRIKOU Dissertação para obtenção do Grau de Mestre em Vinifera EuroMaster – European Master of Sciences of Viticulture and Oenology Orientador: Professor Jorge Ricardo da Silva Júri: Presidente: Olga Laureano, Investigadora Coordenadora, UTL/ISA Vogais: - Antonio Morata, Professor, Universidad Politecnica de Madrid - Jorge Ricardo da Silva, Professor, UTL/ISA Lisboa, 2012 Acknowledgments First and foremost, I would like to thank the Vinifera EuroMaster consortium for giving me the opportunity to participate in the M.Sc. of Viticulture and Enology. Moreover, I would like to express my appreciation to the leading universities and the professors from all around the world for sharing their scientific knowledge and experiences with us and improving day by day the program through mobility. Furthermore, I would like to thank the ISA/UTL University of Lisbon and the personnel working in the laboratory of Enology for providing me with tools, help and a great working environment during the experimental period of this thesis. Special acknowledge to my Professor Jorge Ricardo Da Silva for tutoring me throughout my experiment, but also for the chance to think freely and go deeper to the field of phenols. Last but most important, I would like to extend my special thanks to my family and friends for being a true support and inspiration in every doubt and decision. 1 UTL/ISA University of Lisbon “Vinifera Euromaster” European Master of Science in Viticulture&Oenology Ioanna Mavrikou: Inteferents in condensed tannins quantification with vanillin assay MSc Thesis: 67 pages Key Words: Proanthocyanidins; Interference substances; Phenols; Vanillin assay Abstract Different methods have been established in order to perform accurately the quantification of the condensed tannins in various plant products and beverages. -

EEE M W 24B 24A 27B 27A N Patent Application Publication Dec
US 2009031 1494A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0311494 A1 YAMASHTA et al. (43) Pub. Date: Dec. 17, 2009 (54) RELIEF PRINTING PLATE PRECURSOR FOR (30) Foreign Application Priority Data LASER ENGRAVING, RELIEF PRINTING PLATE, AND PROCESS FOR PRODUCING Jun. 17, 2008 (JP) ................................. 2008-157907 RELEF PRINTING PLATE Feb. 10, 2009 (JP) ................................. 2009-028816 (75) Inventors: Masako YAMASHITA, Publication Classification Shizuoka-ken (JP); Atsushi (51) Int. Cl. SUGASAKI, Shizuoka-ken (JP) B32B 3/00 (2006.01) Correspondence Address: GO3F 7/20 (2006.01) Moss & Burke, PLLC GO3F 7/004 (2006.01) 401 Holland Lane, Suite 407 Alexandria, VA 22314 (US) (52) U.S. Cl. .................... 428/195.1: 430/306: 430/286.1 (73) Assignee: FUJIFILM CORPORATION, (57) ABSTRACT Tokyo (JP) A relief printing plate precursor for laser engraving, including (21) Appl. No.: 12/476,260 a relief forming layer containing (A) a polymerizable com pound having an ethylenic unsaturated bond. (B) a binder (22) Filed: Jun. 2, 2009 polymer, and (C) a compound having deodorizing ability. 11 50 FA - 42 SUB SCANNING DIRECTION -10 - 228 7.s 55 21B EEE m w 24B 24A 27B 27A N Patent Application Publication Dec. 17, 2009 US 2009/0311494 A1 F.G. 1 FA SCANNING DIRECTION 7OA a. CSy ra & 5A - 27WSNS AD 23Ar S3EEASEE21 E-25sagaa EEEEEEEEEEEEEEEEEEEEEEEEE-22s awslighlights fskillsw. 21B 2 TTT "TT". US 2009/031 1494 A1 Dec. 17, 2009 RELEF PRINTING PLATE PRECURSORFOR mask to develop and remove an uncured area, and there is LASER ENGRAVING, RELIEF PRINTING room for improvement since development treatment is nec PLATE, AND PROCESS FOR PRODUCING essary. -

Optimization of the Extraction of Procyanidin B-2 Rich Extract From
Louisiana State University LSU Digital Commons LSU Master's Theses Graduate School 2017 Optimization of the Extraction of Procyanidin B-2 Rich Extract from Unfermented Cocoa Using Response Surface Methodology and Interaction of Procyanidin B-2 Rich Cocoa Extract with Collagenase and Elastase as Biomarkers of Skin Aging Marco Eduardo Toc Sagra Louisiana State University and Agricultural and Mechanical College, [email protected] Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_theses Part of the Life Sciences Commons Recommended Citation Toc Sagra, Marco Eduardo, "Optimization of the Extraction of Procyanidin B-2 Rich Extract from Unfermented Cocoa Using Response Surface Methodology and Interaction of Procyanidin B-2 Rich Cocoa Extract with Collagenase and Elastase as Biomarkers of Skin Aging" (2017). LSU Master's Theses. 4498. https://digitalcommons.lsu.edu/gradschool_theses/4498 This Thesis is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Master's Theses by an authorized graduate school editor of LSU Digital Commons. For more information, please contact [email protected]. OPTIMIZATION OF THE EXTRACTION OF PROCYANIDIN B-2 RICH EXTRACT FROM UNFERMENTED COCOA USING RESPONSE SURFACE METHODOLOGY AND INTERACTION OF PROCYANIDIN B-2 RICH COCOA EXTRACT WITH COLLAGENASE AND ELASTASE AS BIOMARKERS OF SKIN AGING A Thesis Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Master of Science in The School of Nutrition and Food Sciences by Marco Eduardo Toc Sagra B.S., Zamorano University, 2013 August 2017 ACKNOWLEDGMENTS I would like to thank God for always being my inspiration to work hard and with excellence during this stage of my life. -

Transcriptomic and Metabolomic Joint Analysis Reveals Distinct Flavonoid Biosynthesis Regulation for Variegated Testa Color Deve
Transcriptomic and metabolomic joint analysis reveals distinct flavonoid biosynthesis regulation for variegated testa color development in peanut (Arachis hypogaea L.) Mengdie Hu1,Jiawei Li1, Mingyu Hou1,Xiaoqing Liu1,Shunli Cui1,Xinlei Yang1,Lifeng Liu1,Xiaoxia Jiang2, Guojun Mu1* 1.North China Key Laboratory for Crop Germplasm Resources of Education Ministry, College of Agronomy, Hebei Agricultural University, Baoding, China 071001. 2.Hebei Yiyuan Ecological Agriculture Technology Co, Ltd, Baoding, China 074200 [email protected] (M.H.);[email protected] (J.L.);[email protected] (M.H.);[email protected] (X.L.); [email protected] (S.C.);[email protected] (L.Y.);[email protected] (L.L.);[email protected] (X.J.) * Correspondence:[email protected];Tel:+86-1380-326-0389 Abstract: Peanut (Arachis hypogaea L.) is one of the important oil and economic crops, among which the variegated testa peanut is a unique member. But the molecular mechanisms underlying the pigment synthesis in variegated testa are still unclear. Differentially expressed genes (DEGs) in pigment metabolism pathway in colored area indicated there were 27 DEGs highly related to the synthesis of variegated testa color among 1,050 DEGs,which were 13 up-regulated and 14 down-regulated,consisting of 3 PALs, 1 C4H, 2 CHSs, 1 F3H, 1 F3'H, 2 DFRs, 2 LARs, 2 IAAs, 4 bHLHs and 9 MYBs. GO analysis indicated DEGs were similarly enriched in 3 branches.KEGG analysis suggested flavonoid biosynthesis is the most direct metabolic pathway for the synthesis of testa variegation.The liquid chromatography tandem mass spectrometry (LC-MS/MS) results showed that cyanidin and delphinidin were the main metabolites that caused the color difference between the colored area and the non-colored area. -

The Influence of Temperature, Storage Conditions, Ph, and Ionic Strength
molecules Article The Influence of Temperature, Storage Conditions, pH, and Ionic Strength on the Antioxidant Activity and Color Parameters of Rowan Berry Extracts Elena Cristea 1 , Aliona Ghendov-Mosanu 1,* , Antoanela Patras 2 , Carmen Socaciu 3 , Adela Pintea 3 , Cristina Tudor 3 and Rodica Sturza 1 1 Department of Oenology and Chemistry, Food Technology, Faculty Food Technology, Technical University of Moldova, 9/9 Studentilor Street, MD-2045 Chisinau, Moldova; [email protected] (E.C.); [email protected] (R.S.) 2 Department of Sciences, Faculty of Horticulture, “Ion Ionescu de la Brad” Iasi University of Life Sciences, 3 Mihail Sadoveanu Alley, 700490 Iasi, Romania; [email protected] 3 Department of Chemistry and Biochemistry, Faculty of Veterinary Medicine, University of Agricultural Sciences and Veterinary Medicine Cluj-Napoca, Manastur Street, 3–5, 400372 Cluj-Napoca, Romania; [email protected] (C.S.); [email protected] (A.P.); [email protected] (C.T.) * Correspondence: [email protected] Abstract: Recent trends in the food industry combined with novel methods in agriculture could transform rowan into a valuable raw material with potential technological applications. Thus, the aim of this research was to investigate the content of bioactive compounds in its fruits and to Citation: Cristea, E.; assess the color and antioxidant stability of the extracts prepared from such fruits during various Ghendov-Mosanu, A.; Patras, A.; thermal treatments and at different pH and ionic strength values. Various spectrophotometric Socaciu, C.; Pintea, A.; Tudor, C.; methods, HPLC, and capillary electrophoresis were used to quantify the concentrations of bioactive Sturza, R. The Influence of compounds—polyphenols, carotenoids, organic acids, and to assess antioxidant activity and color.