Spatiotemporal Dynamics of Human Microglia Are Linked with Brain Developmental Processes Across the Lifespan
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cell Migration in the Developing Rodent Olfactory System
Cell. Mol. Life Sci. (2016) 73:2467–2490 DOI 10.1007/s00018-016-2172-7 Cellular and Molecular Life Sciences REVIEW Cell migration in the developing rodent olfactory system 1,2 1 Dhananjay Huilgol • Shubha Tole Received: 16 August 2015 / Revised: 8 February 2016 / Accepted: 1 March 2016 / Published online: 18 March 2016 Ó The Author(s) 2016. This article is published with open access at Springerlink.com Abstract The components of the nervous system are Abbreviations assembled in development by the process of cell migration. AEP Anterior entopeduncular area Although the principles of cell migration are conserved AH Anterior hypothalamic nucleus throughout the brain, different subsystems may predomi- AOB Accessory olfactory bulb nantly utilize specific migratory mechanisms, or may aAOB Anterior division, accessory olfactory bulb display unusual features during migration. Examining these pAOB Posterior division, accessory olfactory bulb subsystems offers not only the potential for insights into AON Anterior olfactory nucleus the development of the system, but may also help in aSVZ Anterior sub-ventricular zone understanding disorders arising from aberrant cell migra- BAOT Bed nucleus of accessory olfactory tract tion. The olfactory system is an ancient sensory circuit that BST Bed nucleus of stria terminalis is essential for the survival and reproduction of a species. BSTL Bed nucleus of stria terminalis, lateral The organization of this circuit displays many evolution- division arily conserved features in vertebrates, including molecular BSTM Bed nucleus of stria terminalis, medial mechanisms and complex migratory pathways. In this division review, we describe the elaborate migrations that populate BSTMa Bed nucleus of stria terminalis, medial each component of the olfactory system in rodents and division, anterior portion compare them with those described in the well-studied BSTMpl Bed nucleus of stria terminalis, medial neocortex. -

Visual Impairment in the Absence of Dystroglycan
13136 • The Journal of Neuroscience, October 21, 2009 • 29(42):13136–13146 Neurobiology of Disease Visual Impairment in the Absence of Dystroglycan Jakob S. Satz,1,2,3,4 Alisdair R. Philp,1,6 Huy Nguyen,5 Hajime Kusano,1,2,3,4 Jane Lee,1,2,3,4 Rolf Turk,1,2,3,4 Megan J. Riker,6 Jasmine Herna´ndez,6 Robert M. Weiss,4 Michael G. Anderson,2,6 Robert F. Mullins,6 Steven A. Moore,5 Edwin M. Stone,1,6 and Kevin P. Campbell1,2,3,4 1Howard Hughes Medical Institute and Departments of 2Molecular Physiology and Biophysics, 3Neurology, 4Internal Medicine, 5Pathology, and 6Ophthalmology and Visual Sciences, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa 52242 Ocular involvement in muscular dystrophy ranges from structural defects to abnormal electroretinograms. While the mechanisms underlyingtheabnormalretinalphysiologyinpatientsarenotunderstood,itisthoughtthat␣-dystroglycanextracellularinteractionsare critical for normal visual function. Here we show that -dystroglycan anchors dystrophin and the inward rectifying K ϩ channel Kir4.1 at glial endfeet and that disruption of dystrophin and potassium channel clustering in dystroglycan mutant mice is associated with an attenuationoftheelectroretinogramb-wave.Glial-specificinactivationofdystroglycanordeletionofthecytoplasmicdomainof-dystroglycan was sufficient to attenuate the electroretinogram b-wave. Unexpectedly, deletion of the -dystroglycan cytoplasmic domain did not disrupt the laminar structure of the retina. In contrast to the role of ␣-dystroglycan extracellular interactions during early development of the CNS, -dystroglycan intracellular interactions are important for visual function but not the laminar development of the retina. Introduction and dystrobrevin (Ervasti and Campbell, 1991; Ibraghimov- Muscular dystrophies with ocular involvement are associated Beskrovnaya et al., 1992; Cohn and Campbell, 2000). -

Dynamic Changes in the Localization of Synapse Associated Proteins
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1999 Dynamic changes in the localization of synapse associated proteins during development and differentiation of the mammalian retina Mary Heather West Greenlee Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Cell Biology Commons, Molecular Biology Commons, Neuroscience and Neurobiology Commons, and the Ophthalmology Commons Recommended Citation Greenlee, Mary Heather West, "Dynamic changes in the localization of synapse associated proteins during development and differentiation of the mammalian retina " (1999). Retrospective Theses and Dissertations. 12625. https://lib.dr.iastate.edu/rtd/12625 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly fijom the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter fiice, ^xiule others may be from any type of conq>uter printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. -

ATF5 Deficiency Causes Abnormal Cortical Development
www.nature.com/scientificreports OPEN ATF5 defciency causes abnormal cortical development Mariko Umemura*, Yasuyuki Kaneko, Ryoko Tanabe & Yuji Takahashi Activating transcription factor 5 (ATF5) is a member of the cAMP response element binding protein (CREB)/ATF family of basic leucine zipper transcription factors. We previously reported that ATF5- defcient (ATF5−/−) mice exhibited behavioural abnormalities, including abnormal social interactions, reduced behavioural fexibility, increased anxiety-like behaviours, and hyperactivity in novel environments. ATF5−/− mice may therefore be a useful animal model for psychiatric disorders. ATF5 is highly expressed in the ventricular zone and subventricular zone during cortical development, but its physiological role in higher-order brain structures remains unknown. To investigate the cause of abnormal behaviours exhibited by ATF5−/− mice, we analysed the embryonic cerebral cortex of ATF5−/− mice. The ATF5−/− embryonic cerebral cortex was slightly thinner and had reduced numbers of radial glial cells and neural progenitor cells, compared to a wild-type cerebral cortex. ATF5 defciency also afected the basal processes of radial glial cells, which serve as a scafold for radial migration during cortical development. Further, the radial migration of cortical upper layer neurons was impaired in ATF5−/− mice. These results suggest that ATF5 defciency afects cortical development and radial migration, which may partly contribute to the observed abnormal behaviours. Activating transcription factor 5 (ATF5) is a member of the cAMP response element binding protein (CREB)/ ATF family of basic leucine zipper transcription factors1. Our group and others have reported that ATF5 is a stress responsive transcription factor under conditions such as endoplasmic reticulum (ER) stress and oxidative stress2–4. -

Formation and Function of Neural Circuits Underlying Visual and Locomotor Behaviors Woods Hole Zebrafish Course – August 17, 2013
Visual & Motor Behaviors- 8-17-2013 Formation and function of neural circuits underlying visual and locomotor behaviors Woods Hole Zebrafish Course – August 17, 2013 Instructors for visual behaviors sections: Jim Fadool ([email protected]) John Dowling ([email protected]) Instructors for motor behaviors sections: Michael Granato ( [email protected] ) Roshan Jain ([email protected]) Marc Wolman ([email protected]) Overview: In this last part of the course, we focus on the formation and function of neural circuits underlying visual and locomotor behaviors. It is therefore important to ask: What are the advantages of zebrafish for studies of neuronal circuits and behavior? - the transparency of the embryo and larvae, which has made them so useful in screening for developmental defects, also allows for optical studies of their neuronal circuits. - the simplicity of the nervous system (compared to other vertebrate model systems) and the detailed maps of identified spinal cord and hindbrain neurons allow the study of circuits and behavior at a single cell resolution. -the extrauterine development facilitates a number of powerful techniques, including single cell recording, labeling of individual neurons and their projections, application of drugs, and lesion experiments using sharp needles or a laser beam. - the development of genetic constructs such as channelrhodopsin and halorhodopsin allow for optical activation and silencing of neurons. - the availability of mutants and transgenic lines and the ease with which they can be generated. Together, these represent an unparalleled spectrum of techniques available to study the many different aspects of neural circuits and behavior in a single, intact vertebrate. -

Engineering Region-Specific Brain Organoids from Human Stem Cells to Study Neural Development and Disease
CHAPTER TWELVE Building the brain from scratch: Engineering region-specific brain organoids from human stem cells to study neural development and disease Fadi Jacoba,b,c, Jordan G. Schnolla, Hongjun Songa,d,e,f, and Guo-li Minga,d,e,g,* aDepartment of Neuroscience and Mahoney Institute for Neurosciences, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States bThe Solomon H. Snyder Department of Neuroscience, Johns Hopkins University School of Medicine, Baltimore, MD, United States cMedical Scientist Training Program, Johns Hopkins University School of Medicine, Baltimore, MD, United States dDepartment of Cell and Developmental Biology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States eInstitute for Regenerative Medicine, University of Pennsylvania, Philadelphia, PA, United States fThe Epigenetics Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States gDepartment of Psychiatry, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States *Corresponding author: e-mail address: [email protected] Contents 1. Introduction 478 1.1 Fundamentals of mammalian brain development 479 1.2 Human-specific features 482 1.3 Comparison of in vitro human cell models 482 2. Generation of region-specific brain organoids from human stem cells 484 2.1 Unguided differentiation: Cerebral organoids 484 2.2 Guided differentiation: Region-specific brain organoids 489 3. Advancements in cellular complexity of brain organoids 496 3.1 Fusion of region-specific brain organoids 497 3.2 Enhancing glial cell production and maturation 499 3.3 Reconstitution of resident immune cells and vasculature 500 3.4 Technical modifications for long-term culture 501 3.5 In vivo orthotopic xenotransplantation 503 4. -

Brain-Derived Neurotrophic Factor Participates in Determination of Neuronal Laminar Fate in the Developing Mouse Cerebral Cortex
13218 • The Journal of Neuroscience, December 20, 2006 • 26(51):13218–13230 Development/Plasticity/Repair Brain-Derived Neurotrophic Factor Participates in Determination of Neuronal Laminar Fate in the Developing Mouse Cerebral Cortex Hidefumi Fukumitsu,1 Masanari Ohtsuka,1 Rina Murai,1 Hiroyuki Nakamura,2 Kazuo Itoh,2 Shoei Furukawa1 1Laboratory of Molecular Biology, Gifu Pharmaceutical University, Gifu 502-8585, Japan, and 2Department of Morphological Neuroscience, Gifu University Graduate School of Medical Science, Gifu 501-1194, Japan Lamina formation in the developing cerebral cortex requires precisely regulated generation and migration of the cortical progenitor cells. To test the possible involvement of brain-derived neurotrophic factor (BDNF) in the formation of the cortical lamina, we investigated the effects of BDNF protein and anti-BDNF antibody separately administered into the telencephalic ventricular space of 13.5-d-old mouse embryos. BDNF altered the position, gene-expression properties, and projections of neurons otherwise destined for layer IV to those of neurons for the deeper layers, V and VI, of the cerebral cortex, whereas anti-BDNF antibody changed some of those of neurons of upper layers II/III. Additional analysis revealed that BDNF altered the laminar fate of neurons only if their parent progenitor cells were exposed to it at approximately S-phase and that it hastened the timing of the withdrawal of their daughter neurons from the ventricular prolifer- ating pool by accelerating the completion of S-phase, downregulation of the Pax6 (paired box gene 6) expression, an essential transcrip- tion factor for generation of the upper layer neurons, and interkinetic nuclear migration of cortical progenitors in the ventricular zone. -

Isolation of an Allele of Reeler by Insertional Mutagenesis GRAHAM G
Proc. Nati. Acad. Sci. USA Vol. 91, pp. 11050-11054, November 1994 Genetics Isolation of an allele of reeler by insertional mutagenesis GRAHAM G. MIAO*t, RICHARD J. SMEYNE*, GABRIELLA D'ARCANGELO*, NEAL G. COPELAND§, NANCY A. JENKINS§, JAMES I. MORGAN*, AND TOM CURRAN*¶ *Roche Institute of Molecular Biology, Nutley, NJ 07110; tDepartment of Biological Sciences, Columbia University, New York, NY 10027; tNeurogenetics Unit, Department of Central Nervous System Research, Hoffmann-La Roche, Inc., Nutley, NJ 07110; and §Mammalian Genetics Laboratory, Advanced BioScience Laboratories-Basic Research Program, National Cancer Institute-Frederick Cancer Research and Development Center, Frederick, MD 21702 Communicated by Herbert Weissbach, July 12, 1994 ABSTRACT Reeler (rl) Is an autosomal recessive mutation the rl locus. Thus, a DNA marker has been identified that can that affects migration of postmitotic neurons in the mouse now be used to identify and isolate the rl gene. central nervous system. The reeler (ri/rl) mouse displays a disruption oflaminar structures in both the cerebellum and the MATERIALS AND METHODS forebrain and it exhibits tremors, dystonia, and ataxia. The molecular basis ofthe reeler phenotype is unknown because the Transgene and Transgenic Mice. The sf transgene was gene involved has not yet been identified. We report here the generated by inserting a 12-nt sequence (CCGCTC- isolation and characterization of an allele of rl, reelerlmPe GAGCGG) containing a Xho I site between nt 1922 and nt (rit). This allele was generated by the fortuitous insertion of a 1923 in the third exon of the mouse c-fos (12). A 4.7-kb sf transgene, supfos (sf), into the mouse rllocus. -
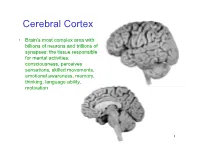
Cerebral Cortex
Cerebral Cortex • Brain’s most complex area with billions of neurons and trillions of synapses: the tissue responsible for mental activities: consciousness, perceives sensations, skilled movements, emotional awareness, memory, thinking, language ability, motivation 1 John Lorber, Science 210:1232 (1980) “There a young student at this university who has an IQ of 126, has gained a first-class honors degree in mathematics, and is socially completely normal. And yet the boy has virtually no brain.” “The student’s physician at the university noticed that the student had slightly larger than normal head… When we did a brain scan on him we saw that instead of the normal 4.5 cm thickness of brain tissue there was just a millimeter or so. His cranium is filled with CSF.” How is this possible? What does it tell us? Do you think this would be OK if it happened to an adult? To a 15 year old? To a 5 year old? To a neonate? 3 Types of Cerebral Cortex • Neocortex – Newest in evolution – About 90% of total – 6 layers, most complex • Paleocortex – Associated with olfactory system, the parahippocampal gyrus, uncus – fewer than 6 layers • Archicortex – Hippocampal formation; limbic system – 3 layers, most primitive • Mesocortex – Cingulate gyrus, insular cortex – Transitional between archicortex and neocortex 5 The perks of having a neocortex • The words used to describe the higher mental capacities of animals with a large neocortex include: – CONSCIOUSNESS – FREE WILL – INTELLIGENCE – INSIGHT • Animals with much simpler brains learn well, so LEARNING should not be among these capacities (Macphail 1982). • A species could have genetically determined mechanisms, acquired through evolutionary selection, for taking advantage of the regular features of the environment, or they could have learned through direct experience. -

Oxford Handbooks Online
Fetal and Infant Neurobehavioral Development Oxford Handbooks Online Fetal and Infant Neurobehavioral Development: Basic Processes and Environmental Influences Catherine Monk and Amie Ashley Hane The Oxford Handbook of Perinatal Psychology (Forthcoming) Edited by Amy Wenzel Subject: Psychology, Health Psychology, Clinical Psychology Online Publication Date: Oct DOI: 10.1093/oxfordhb/9780199778072.013.20 2014 Abstract and Keywords This chapter reviews the literature examining fetal and infant neurobehavioral development. Basic fetal neurological development and neurobehavioral functioning are reviewed. Major fetal neurobehavioral milestones and their assessment are addressed and include fetal behavioral states, heart rate, movement, and responsivity to stimuli. The processes of neurological growth from birth to age 2 are reviewed. Infant neurobehavioral development is addressed and includes state regulation and sleep, physical growth and motor development, and the basic processes underlying social-emotional development. For fetus and infant, research examining the associations between neurobehavioral development and maternal distress and poverty is reviewed. The implications for future directions in fetal-infant neurobehavioral research are discussed. Keywords: Prenatal development, fetus, infant, mother-infant, neurological development, physiology This chapter on fetal and infant neurobehavioral development covers both periods with nearly equal weight, reflecting, in part, the greater accessibility of the fetus to research and, more significantly, the increasing emphasis on developmental roots for later functioning, including those that take hold during fetal development. For both the fetus and infant, we cover basic brain development and key neurobehavioral milestones, some of which overlap conceptually (i.e., state regulation), as well as micro- and macrolevel contextual factors that appear to influence the developmental trajectories at each time: maternal distress and poverty. -

The Laminar Organization of the Lateral Geniculate Body and the Striate Cortex in the Squirrel Monkey (Saimiri Sciureus)’
0270.6474/83/0304-0673$02.00/O The Journal of Neuroscience Copyright 0 Society for Neuroscience Vol. 3, No. 4, pp. 673-702 Printed in U.S.A. April 1983 THE LAMINAR ORGANIZATION OF THE LATERAL GENICULATE BODY AND THE STRIATE CORTEX IN THE SQUIRREL MONKEY (SAIMIRI SCIUREUS)’ DAVID FITZPATRICK,” KAZUO ITOH,:’ AND IRVING T. DIAMOND” Department of Psychology, Duke University, Durham, North Carolina 27706 Received June 4, 1982; Revised October 20, 1982; Accepted October 21, 1982 Abstract The organization of the projection from the lateral geniculate body to the striate cortex in the squirrel monkey has been re-examined using the anterograde and retrograde transport of horseradish peroxidase (HRP) and wheat germ agglutinin conjugated to HRP. The results confirm earlier findings that the projections of the magnocellular and parvocellular layers of the lateral geniculate body terminate in separate sublaminae of layer IV of striate cortex; a more superficial projection of the parvocellular layers to a narrow strip at the base of layer III (IVA in Brodmann’s terminology) has also been confirmed. In addition to these well characterized pathways, our results show that the projections of the lateral geniculate body terminate in more superficial levels of layer III and sparsely in layer I of striate cortex. The projections to the upper portion of layer III terminate in distinct patches which coincide precisely with patches of cytochrome oxidase activity previously identified in this zone. The projections to the patches originate primarily from small, pale-staining cells of the “intercalated layers” which surround the magnocellular layers of the lateral geniculate body. -

An Autoradiographic Study of the Ontogeny of Receptor
AN AUTORADIOGRAPHIC STUDY OF THE ONTOGENY OF RECEPTOR BINDING SITES IN THE CENTRAL VISUAL PATHWAY OF THE RAT. Dominic Hansel Lobo Submitted for the Degree of Doctor of Philosophy Department of Anatomy and Developmental Neurobiology University College, London MAY 1991 1 ProQuest Number: 10629656 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 10629656 Published by ProQuest LLC(2017). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 TO ALL MY NDUGU’S 2 I ACKNOWLEDGMENTS First and foremost, this is dedicated to the three wise men who have shown me the True Way, Professor Guz for being my usurer, to Ndugu for his concern in my affairs, the Family for all the inspiration and, to Dooz for seeing me over the final hurdles and sorting out the final edit. My thanks and appreciation extends to Dr. John Pamavelas for his faith in me through all the trials and tribulations, Professor Lieberman for the use of laboratory space, Dr. Pradeep Bhide for his exorbitant consultation fees, Dr. Mike Stewart for the use of the Image Analysis system at the Open University, Dr.