Genetics of Long-QT Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Non Commercial Use Only
Cardiogenetics 2017; volume 7:6304 Sudden death in a young patient with atrial fibrillation Case Report Correspondence: María Angeles Espinosa Castro, Inherited Cardiovascular Disease A 22-year-old man suffered a sudden Program, Cardiology Department, Gregorio María Tamargo, cardiac arrest without previous symptoms Marañón Hospital, Dr. Esquerdo, 46, 28007, María Ángeles Espinosa, while he was at rest, waiting for a subway Madrid, Spain. Víctor Gómez-Carrillo, Miriam Juárez, train. Cardiopulmonary resuscitation was Tel.: +34.91.586.82.90. immediately started using an Automated E-mail: [email protected] Francisco Fernández-Avilés, External Defibrillation that identified the Raquel Yotti Key words: KCNQ1; mutation; channelopa- presence of ventricular fibrillation and thy; sudden cardiac death; atrial fibrillation. Inherited Cardiovascular Disease delivered a shock. Return of spontaneous Program, Cardiology Department, circulation was achieved after three Contributions: MT, acquisition and interpreta- Gregorio Marañón Hospital, Madrid, attempts, being atrial fibrillation (AF) the tion of data for the work, ensuring that ques- Spain patient’s rhythm at this point (Figure 1). tions related to the accuracy or integrity of any He was admitted to our Cardiovascular part of the work is appropriately investigated Intensive Care Unit and therapeutic and resolved; MAE, conception of the work, hypothermia was performed over a period critical revision of the intellectual content, final approval of the version to be published, Abstract of 24 h. After completing hypothermia, ensuring that questions related to the accuracy rewarming, and another 24 h of controlled of any part of the work is appropriately inves- Sudden cardiac death (SCD) in young normothermia the patient awakened with no tigated and resolved; VG-C, acquisition and patients without structural heart disease is residual neurologic damage. -

Constrictive Pericarditis Causing Ventricular Tachycardia.Pdf
EP CASE REPORT ....................................................................................................................................................... A visually striking calcific band causing monomorphic ventricular tachycardia as a first presentation of constrictive pericarditis Kian Sabzevari 1*, Eva Sammut2, and Palash Barman1 1Bristol Heart Institute, UH Bristol NHS Trust UK, UK; and 2Bristol Heart Institute, UH Bristol NHS Trust UK & University of Bristol, UK * Corresponding author. Tel: 447794900287; fax: 441173425926. E-mail address: [email protected] Introduction Constrictive pericarditis (CP) is a rare condition caused by thickening and stiffening of the pericar- dium manifesting in dia- stolic dysfunction and enhanced interventricu- lar dependence. In the developed world, most cases are idiopathic or are associated with pre- vious cardiac surgery or irradiation. Tuberculosis remains a leading cause in developing areas.1 Most commonly, CP presents with symptoms of heart failure and chest discomfort. Atrial arrhythmias have been described as a rare pre- sentation, but arrhyth- mias of ventricular origin have not been reported. Figure 1 (A) The 12 lead electrocardiogram during sustained ventricular tachycardia is shown; (B and C) Case report Different projections of three-dimensional reconstructions of cardiac computed tomography demonstrating a A 49-year-old man with a striking band of calcification around the annulus; (D) Carto 3DVR mapping—the left hand panel (i) demonstrates a background of diabetes, sinus beat with late potentials at the point of ablation in the coronary sinus, the right hand panel (iii) shows the hypertension, and hyper- pacemap with a 89% match to the clinical tachycardia [matching the morphology seen on 12 lead ECG (A)], and cholesterolaemia and a the middle panel (ii) displays the three-dimensional voltage map. -
Long QT Syndrome Testing
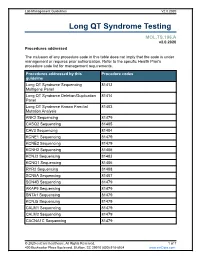
Lab Management Guidelines V2.0.2020 Long QT Syndrome Testing MOL.TS.196.A v2.0.2020 Procedures addressed The inclusion of any procedure code in this table does not imply that the code is under management or requires prior authorization. Refer to the specific Health Plan's procedure code list for management requirements. Procedures addressed by this Procedure codes guideline Long QT Syndrome Sequencing 81413 Multigene Panel Long QT Syndrome Deletion/Duplication 81414 Panel Long QT Syndrome Known Familial 81403 Mutation Analysis ANK2 Sequencing 81479 CASQ2 Sequencing 81405 CAV3 Sequencing 81404 KCNE1 Sequencing 81479 KCNE2 Sequencing 81479 KCNH2 Sequencing 81406 KCNJ2 Sequencing 81403 KCNQ1 Sequencing 81406 RYR2 Sequencing 81408 SCN5A Sequencing 81407 SCN4B Sequencing 81479 AKAP9 Sequencing 81479 SNTA1 Sequencing 81479 KCNJ5 Sequencing 81479 CALM1 Sequencing 81479 CALM2 Sequencing 81479 CACNA1C Sequencing 81479 © 2020 eviCore healthcare. All Rights Reserved. 1 of 7 400 Buckwalter Place Boulevard, Bluffton, SC 29910 (800) 918-8924 www.eviCore.com Lab Management Guidelines V2.0.2020 What is Long QT syndrome Definition Long QT Syndrome (LQTS) is caused by mutations in a number of genes, most of which are related to the functioning of sodium or potassium ion channels in the heart.1 Testing may offer prognostic information in some cases, as specific genes and even specific mutations within those genes may have some correlation to risk for sudden death, effectiveness of beta-blocker therapy, and preventive strategies.1-4 Signs and symptoms of long QT syndrome (LQTS) are variable, but may include a prolonged QT interval on an electrocardiogram, torsades de pointes, syncope, seizures, cardiac arrest, and sudden cardiac death.1,2 Many patients with LQTS can be largely asymptomatic, with cardiac arrest or sudden cardiac death as the first and only symptom. -

What Are Premature Ventricular Contractions?
What Are Premature Ventricular Contractions? Premature ventricular contractions (VPCs or PVCs) are irregular beats that originate from the heart’s pumping chambers (ventricles). These beats interrupt the normal regular (sinus) rhythm, and result in irregularity to the heart rhythm. Although single PVCs are not life- threatening, they may indicate the presence of underlying heart disease, or when they become more severe can cause rapid and dangerous increases in heart rate (ventricular tachycardia). WHAT CAUSES PREMATURE VENTRICULAR CONTRACTIONS? PVCs can occur secondary to a variety of causes, most concerning being cardiac disease. PVCs are most commonly seen in dogs with Dilated Cardiomyopathy, but can also occur in the later stages of disease with Myxomatous Mitral Valve Degeneration. PVCs and other ventricular arrhythmias can also with no evidence of underlying structural heart disease. The workup for PVCs can be frustrating, because PVCs can also occur secondary to conditions unrelated to the heart. Most commonly they can occur secondary to gastrointestinal disease, systemic disease, and pain. HOW ARE PREMATURE VENTRICULAR CONTRACTIONS DIAGNOSED? Most commonly, your veterinarian will hear an irregular heart rhythm and recommend an electrocardiogram (ECG) to assess the heart rhythm. The ECG will enable your veterinarian to determine whether the irregular rhythm is due to PVCs. Following this diagnosis, there are several additional recommended tests. TESTING Following this diagnosis, there are several additional recommended tests. ECHOCARDIOGRAPHY Echocardiography (heart ultrasound) is recommended to make sure that there is no evidence of structural cardiac disease causing the abnormal heart rhythm. 24-HOUR HOLTER Single PVCs usually do not require treatment, however a Holter monitor will determine whether the frequency or severity of the MONITOR PVCs warrant anti-arrhythmic medication, and make sure that there is no evidence of ventricular tachycardia, which is a life threatening heart rhythm. -

Drugs and Life-Threatening Ventricular Arrhythmia Risk: Results from the DARE Study Cohort
Open Access Research BMJ Open: first published as 10.1136/bmjopen-2017-016627 on 16 October 2017. Downloaded from Drugs and life-threatening ventricular arrhythmia risk: results from the DARE study cohort Abigail L Coughtrie,1,2 Elijah R Behr,3,4 Deborah Layton,1,2 Vanessa Marshall,1 A John Camm,3,4,5 Saad A W Shakir1,2 To cite: Coughtrie AL, Behr ER, ABSTRACT Strengths and limitations of this study Layton D, et al. Drugs and Objectives To establish a unique sample of proarrhythmia life-threatening ventricular cases, determine the characteristics of cases and estimate ► The Drug-induced Arrhythmia Risk Evaluation study arrhythmia risk: results from the the contribution of individual drugs to the incidence of DARE study cohort. BMJ Open has allowed the development of a cohort of cases of proarrhythmia within these cases. 2017;7:e016627. doi:10.1136/ proarrhythmia. Setting Suspected proarrhythmia cases were referred bmjopen-2017-016627 ► These cases have provided crucial safety by cardiologists across England between 2003 and 2011. information, as well as underlying clinical and ► Prepublication history for Information on demography, symptoms, prior medical and genetic data. this paper is available online. drug histories and data from hospital notes were collected. ► Only patients who did not die as a result of the To view these files please visit Participants Two expert cardiologists reviewed data the journal online (http:// dx. doi. proarrhythmia could be included. for 293 referred cases: 130 were included. Inclusion org/ 10. 1136/ bmjopen- 2017- ► Referral of cases by cardiologists alone may have criteria were new onset or exacerbation of pre-existing 016627). -

Atrial Fibrillation (ATRIA) Study
European Journal of Human Genetics (2014) 22, 297–306 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg REVIEW Atrial fibrillation: the role of common and rare genetic variants Morten S Olesen*,1,2,4, Morten W Nielsen1,2,4, Stig Haunsø1,2,3 and Jesper H Svendsen1,2,3 Atrial fibrillation (AF) is the most common cardiac arrhythmia affecting 1–2% of the general population. A number of studies have demonstrated that AF, and in particular lone AF, has a substantial genetic component. Monogenic mutations in lone and familial AF, although rare, have been recognized for many years. Presently, mutations in 25 genes have been associated with AF. However, the complexity of monogenic AF is illustrated by the recent finding that both gain- and loss-of-function mutations in the same gene can cause AF. Genome-wide association studies (GWAS) have indicated that common single-nucleotide polymorphisms (SNPs) have a role in the development of AF. Following the first GWAS discovering the association between PITX2 and AF, several new GWAS reports have identified SNPs associated with susceptibility of AF. To date, nine SNPs have been associated with AF. The exact biological pathways involving these SNPs and the development of AF are now starting to be elucidated. Since the first GWAS, the number of papers concerning the genetic basis of AF has increased drastically and the majority of these papers are for the first time included in a review. In this review, we discuss the genetic basis of AF and the role of both common and rare genetic variants in the susceptibility of developing AF. -

Basic Rhythm Recognition
Electrocardiographic Interpretation Basic Rhythm Recognition William Brady, MD Department of Emergency Medicine Cardiac Rhythms Anatomy of a Rhythm Strip A Review of the Electrical System Intrinsic Pacemakers Cells These cells have property known as “Automaticity”— means they can spontaneously depolarize. Sinus Node Primary pacemaker Fires at a rate of 60-100 bpm AV Junction Fires at a rate of 40-60 bpm Ventricular (Purkinje Fibers) Less than 40 bpm What’s Normal P Wave Atrial Depolarization PR Interval (Normal 0.12-0.20) Beginning of the P to onset of QRS QRS Ventricular Depolarization QRS Interval (Normal <0.10) Period (or length of time) it takes for the ventricles to depolarize The Key to Success… …A systematic approach! Rate Rhythm P Waves PR Interval P and QRS Correlation QRS Rate Pacemaker A rather ill patient……… Very apparent inferolateral STEMI……with less apparent complete heart block RATE . Fast vs Slow . QRS Width Narrow QRS Wide QRS Narrow QRS Wide QRS Tachycardia Tachycardia Bradycardia Bradycardia Regular Irregular Regular Irregular Sinus Brady Idioventricular A-Fib / Flutter Bradycardia w/ BBB Sinus Tach A-Fib VT PVT Junctional 2 AVB / II PSVT A-Flutter SVT aberrant A-Fib 1 AVB 3 AVB A-Flutter MAT 2 AVB / I or II PAT PAT 3 AVB ST PAC / PVC Stability Hypotension / hypoperfusion Altered mental status Chest pain – Coronary ischemic Dyspnea – Pulmonary edema Sinus Rhythm Sinus Rhythm P Wave PR Interval QRS Rate Rhythm Pacemaker Comment . Before . Constant, . Rate 60-100 . Regular . SA Node Upright in each QRS regular . Interval =/< leads I, II, . Look . Interval .12- .10 & III alike .20 Conduction Image reference: Cardionetics/ http://www.cardionetics.com/docs/healthcr/ecg/arrhy/0100_bd.htm Sinus Pause A delay of activation within the atria for a period between 1.7 and 3 seconds A palpitation is likely to be felt by the patient as the sinus beat following the pause may be a heavy beat. -

PAROXYSMAL VENTRICULAR TACHYCARDIA OCCURRING in a NORMAL HEART by DAVID ROMNEY, M.B., B.Ch.(Dub.) Ex-Senior House Officer in Medicine, St
Postgrad Med J: first published as 10.1136/pgmj.31.354.191 on 1 April 1955. Downloaded from I9I -J PAROXYSMAL VENTRICULAR TACHYCARDIA OCCURRING IN A NORMAL HEART By DAVID ROMNEY, M.B., B.Ch.(Dub.) Ex-Senior House Officer in Medicine, St. James's Hospital, Balham It is widely taught-and rightly so-that by sinus pressure will, of course, not affect the paroxysmal ventricular tachycardia is one of the rate in ventricular tachycardia. rarer arrhythmias, and is associated with grave In I953 Froment, Gallavardin and Cahen myocardial damage, with special reference to myo- offered a classification of various forms of cardial infarction. It is the least common, and paroxysmal ventricular tachycardia, and included most serious of the paroxysmal tachycardias, some case report. They described the following which contain four varieties of arrhythmia: supra- groups:- ventricular (auricular and nodal) 60 per cent.; (i) Terminal prefibrillatory ventricular tachy- auricular fibrillation, 30 per cent.; auricular flutter, cardia. 6 per cent.; and ventricular tachycardia, 4 per (ii) Curable and mild monomorphic extra- cent. systoles, with paroxysms of tachycardia. Figures from various sources (Campbell, 1947) (iii) Paroxysmal ventricular tachycardia due toby copyright. would indicate that in the latter group, four-fifths a lesion of the ventricular septum. of the cases had seriously damaged hearts. In (iv) Persistent and prolonged ventricular tachy- the remaining fifth no cause was found for the cardia developing in sound hearts, usually paroxysms. Paroxysmal ventricular tachycardia is in young subjects. more common in men than in women in the proportion of about 3.2. This arrhythmia was Case Report first identified by Sir Thomas Lewis in I909 and A married woman, aged 48, first became aware Gallavardin (1920, I921, 1922) emphasized the in 1948 of a paroxysm which caused alarm, faint- seriousness of the 'terminal ven- ness and It lasted a short pre-fibrillatory collapse. -

Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives?
The Science Journal of the Lander College of Arts and Sciences Volume 11 Number 1 Fall 2017 - 2017 Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives? Menachem Braun Touro College Follow this and additional works at: https://touroscholar.touro.edu/sjlcas Part of the Cardiovascular Diseases Commons, and the Medical Genetics Commons Recommended Citation Braun, M. (2017). Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives?. The Science Journal of the Lander College of Arts and Sciences, 11(1). Retrieved from https://touroscholar.touro.edu/sjlcas/vol11/iss1/8 This Article is brought to you for free and open access by the Lander College of Arts and Sciences at Touro Scholar. It has been accepted for inclusion in The Science Journal of the Lander College of Arts and Sciences by an authorized editor of Touro Scholar. For more information, please contact [email protected]. Should Genetic Testing be Recommended for Long QT Syndrome Patients and Their Relatives? Menachem Braun Menachem Braun graduated with a BS in Biology in September 2017. Abstract 7KH/RQJ476\QGURPH /476 LVDIDPLOLDOSRWHQWLDOO\IDWDOFDUGLDFDUUK\WKPLD7UDGLWLRQDOO\LWKDVEHHQGLDJQRVHGE\(&* Molecular studies have provided evidence that LQTS can be caused by a range of underlying molecular abnormalities. Genetic research has proven that different forms of LQTS have different genotypic bases. Therefore, it has become possible to diagnose WKHVSHFLÀFW\SHRIGLVHDVHJHQHWLFDOO\7KLVVWXG\H[DPLQHVWKHDGYDQFHPHQWVPDGHLQWKHSDVWWKLUW\\HDUVLQXQGHUVWDQGLQJ /476DQGUHVHDUFKUHJDUGLQJWKHXVHRIJHQHWLFWHVWLQJLQRUGHUWRGHWHUPLQHWKHEHQHÀWVRIJHQHWLFWHVWLQJIRUWKLVGLVHDVH$ survey of original studies which produced the information is presented here, and provides the reader with an understanding of the PHFKDQLFVRIWKHGLVHDVHDQGKRZWKH\GLIIHULQWKHVHYHUDOJHQHWLFYDULDQWV5HVHDUFKVKRZVWKDWWKHEHQHÀWRIJHQHWLFWHVWLQJ must be weighed against the personal implications in may have for a particular patient and his or her family. -

Latest Diagnostic and Treatment Strategies for the Congenital Long QT Syndromes
Latest Diagnostic and Treatment Strategies for the Congenital Long QT Syndromes Michael J. Ackerman, MD, PhD Windland Smith Rice Cardiovascular Genomics Research Professor Professor of Medicine, Pediatrics, and Pharmacology Director, Long QT Syndrome Clinic and the Mayo Clinic Windland Smith Rice Sudden Death Genomics Laboratory President, Sudden Arrhythmia Death Syndromes (SADS) Foundation Learning Objectives to Disclose: • To RECOGNIZE the “faces” (phenotypes) of the congenital long QT syndromes (LQTS) • To CRITIQUE the various diagnostic modalities used in the evaluation of LQTS and UNDERSTAND their limitations • To ASSESS the currently available treatment options for the various LQT syndromes and EVALUATE their efficacy WINDLAND Smith Rice Sudden Death Genomics Laboratory Conflicts of Interest to Disclose: • Consultant – Boston Scientific, Gilead Sciences, Medtronic, St. Jude Medical, and Transgenomic/FAMILION • Royalties – Transgenomic/FAMILION Congenital Long QT Syndrome Normal QT interval QT QT Prolonged QT 1. Syncope 2. Seizures 3. Sudden death Torsades de pointes Congenital Long QT Syndrome Normal QT interval QT QT ♥ 1957 – first clinical description – JLNS ♥ 1960s – RomanoProlonged-Ward QT syndrome ♥ 1983 – “Schwartz/Moss score”1. Syncope ♥ 1991 – first LQTS chromosome locus 2. Seizures ♥ March 10, 1995 – birth of cardiac 3. Sudden channelopathies death Torsades de pointes Congenital Long QT Syndrome Normal QT interval QT QT Prolonged QT 1. Syncope 2. Seizures 3. Sudden death Torsades de pointes Congenital Long QT Syndrome Normal -

The Example of Short QT Syndrome Jules C
Hancox et al. Journal of Congenital Cardiology (2019) 3:3 Journal of https://doi.org/10.1186/s40949-019-0024-7 Congenital Cardiology REVIEW Open Access Learning from studying very rare cardiac conditions: the example of short QT syndrome Jules C. Hancox1,4* , Dominic G. Whittaker2,3, Henggui Zhang4 and Alan G. Stuart5,6 Abstract Background: Some congenital heart conditions are very rare. In a climate of limited resources, a viewpoint could be advanced that identifying diagnostic criteria for such conditions and, through empiricism, effective treatments should suffice and that extensive mechanistic research is unnecessary. Taking the rare but dangerous short QT syndrome (SQTS) as an example, this article makes the case for the imperative to study such rare conditions, highlighting that this yields substantial and sometimes unanticipated benefits. Genetic forms of SQTS are rare, but the condition may be under-diagnosed and carries a risk of sudden death. Genotyping of SQTS patients has led to identification of clear ion channel/transporter culprits in < 30% of cases, highlighting a role for as yet unidentified modulators of repolarization. For example, recent exome sequencing in SQTS has identified SLC4A3 as a novel modifier of ventricular repolarization. The need to distinguish “healthy” from “unhealthy” short QT intervals has led to a search for additional markers of arrhythmia risk. Some overlap may exist between SQTS, Brugada Syndrome, early repolarization and sinus bradycardia. Genotype-phenotype studies have led to identification of arrhythmia substrates and both realistic and theoretical pharmacological approaches for particular forms of SQTS. In turn this has increased understanding of underlying cardiac ion channels. -

Ventricular Tachycardia Drugs Versus Devices John Camm St
Cardiology Update 2015 Davos, Switzerland: 8-12th February 2015 Ventricular Arrhythmias Ventricular Tachycardia Drugs versus Devices John Camm St. George’s University of London, UK Imperial College, London, UK Declaration of Interests Chairman: NICE Guidelines on AF, 2006; ESC Guidelines on Atrial Fibrillation, 2010 and Update, 2012; ACC/AHA/ESC Guidelines on VAs and SCD; 2006; NICE Guidelines on ACS and NSTEMI, 2012; NICE Guidelines on heart failure, 2008; NICE Guidelines on Atrial Fibrillation, 2006; ESC VA and SCD Guidelines, 2015 Steering Committees: multiple trials including novel anticoagulants DSMBs: multiple trials including BEAUTIFUL, SHIFT, SIGNIFY, AVERROES, CASTLE- AF, STAR-AF II, INOVATE, and others Events Committees: one trial of novel oral anticoagulants and multiple trials of miscellaneous agents with CV adverse effects Editorial Role: Editor-in-Chief, EP-Europace and Clinical Cardiology; Editor, European Textbook of Cardiology, European Heart Journal, Electrophysiology of the Heart, and Evidence Based Cardiology Consultant/Advisor/Speaker: Astellas, Astra Zeneca, ChanRX, Gilead, Merck, Menarini, Otsuka, Sanofi, Servier, Xention, Bayer, Boehringer Ingelheim, Bristol- Myers Squibb, Daiichi Sankyo, Pfizer, Boston Scientific, Biotronik, Medtronic, St. Jude Medical, Actelion, GlaxoSmithKline, InfoBionic, Incarda, Johnson and Johnson, Mitsubishi, Novartis, Takeda Therapy for Ventricular Tachycardia Medical therapy Antiarrhythmic drugs Autonomic management Ventricular tachycardia Monomorphic Polymorphic Ventricular fibrillation Ventricular storms Ablation therapy Device therapy Surgical Defibrillation Catheter Antitachycardia pacing History of Antiarrhythmic Drugs 1914 - Quinidine 1950 - Lidocaine 1951 - Procainamide 1946 – Digitalis 1956 – Ajmaline 1962 - Verapamil 1962 – Disopyramide 1964 - Propranolol 1967 – Amiodarone 1965 – Bretylium 1972 – Mexiletine 1973 – Aprindine, Tocainide 1969 - Diltiazem 1975- Flecainide 1976 – Propafenone Encainide Ethmozine 2000 - Sotalol D-sotalol 1995 - Ibutilide (US) Recainam 2000 – Dofetilide US) IndecainideX Etc.