Non Commercial Use Only
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
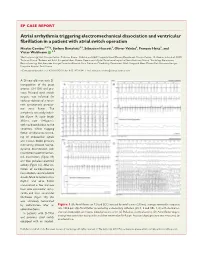
Atrial Arrhythmia Triggering Electromechanical Dissociation And
EP CASE REPORT ....................................................................................................................................................... Atrial arrhythmia triggering electromechanical dissociation and ventricular fibrillation in a patient with atrial switch operation Nicolas Combes1,2,3*, Stefano Bartoletti1,2,Se´bastien Hascoet€ 3, Olivier Vahdat2, Franc¸ois Heitz2, and Victor Waldmann 4,5 1Electrophysiology Unit, Clinique Pasteur, Toulouse, France; 2Pediatric and Adult Congenital Heart Disease Department, Clinique Pasteur, 45, Avenue de Lombez, 31076 Toulouse, France; 3Pediatric and Adult Congenital Heart Disease Department, Hoˆpital Marie Lannelongue, Le Plessis-Robinson, France; 4Cardiology Department, Electrophysiology Unit, European Georges Pompidou Hospital, Paris, France; and 5Cardiology Department, Adult Congenital Heart Disease Unit, European Georges Pompidou Hospital, Paris, France * Corresponding author. Tel: 133 562213131; fax: 133 562211641. E-mail address: [email protected] A 26-year-old man with D- transposition of the great arteries (D-TGA) and pre- vious Mustard atrial switch surgery was referred for catheter ablation of a recur- rent symptomatic paroxys- mal atrial flutter. The arrhythmia was easily induci- ble (Figure 1A, cycle length 310 ms, rate 194 b.p.m.), with rapid conduction to the ventricles. While mapping flutter, simultaneous record- ing of endocardial signals and invasive blood pressure monitoring showed haemo- dynamic deterioration with intermittent electromechan- ical dissociation (Figure 1B) and then pulseless electrical activity (Figure 1C). After ini- tiation of cardiopulmonary resuscitation, several electric shocks failed to restore sinus rhythm and atrial flutter transitioned a few minutes later into ventricular tachy- cardia and then ventricular fibrillation (Figure 1D); this was ultimately terminated by defibrillation after an Figure 1 (A) Atrial flutter on 12-lead ECG induced by atrial bursts (240 ms), average ventricular response adrenaline bolus. -

Mitral Valve Prolapse, Arrhythmias, and Sudden Cardiac Death: the Role of Multimodality Imaging to Detect High-Risk Features
diagnostics Review Mitral Valve Prolapse, Arrhythmias, and Sudden Cardiac Death: The Role of Multimodality Imaging to Detect High-Risk Features Anna Giulia Pavon 1,2,*, Pierre Monney 1,2,3 and Juerg Schwitter 1,2,3 1 Cardiac MR Center (CRMC), Lausanne University Hospital (CHUV), 1100 Lausanne, Switzerland; [email protected] (P.M.); [email protected] (J.S.) 2 Cardiovascular Department, Division of Cardiology, Lausanne University Hospital (CHUV), 1100 Lausanne, Switzerland 3 Faculty of Biology and Medicine, University of Lausanne (UniL), 1100 Lausanne, Switzerland * Correspondence: [email protected]; Tel.: +41-775-566-983 Abstract: Mitral valve prolapse (MVP) was first described in the 1960s, and it is usually a benign condition. However, a subtype of patients are known to have a higher incidence of ventricular arrhythmias and sudden cardiac death, the so called “arrhythmic MVP.” In recent years, several studies have been published to identify the most important clinical features to distinguish the benign form from the potentially lethal one in order to personalize patient’s treatment and follow-up. In this review, we specifically focused on red flags for increased arrhythmic risk to whom the cardiologist must be aware of while performing a cardiovascular imaging evaluation in patients with MVP. Keywords: mitral valve prolapse; arrhythmias; cardiovascular magnetic resonance Citation: Pavon, A.G.; Monney, P.; Schwitter, J. Mitral Valve Prolapse, Arrhythmias, and Sudden Cardiac Death: The Role of Multimodality 1. Mitral Valve and Arrhythmias: A Long Story Short Imaging to Detect High-Risk Features. In the recent years, the scientific community has begun to pay increasing attention Diagnostics 2021, 11, 683. -

Antithrombotic Therapy in Atrial Fibrillation Associated with Valvular Heart Disease
Europace (2017) 0, 1–21 EHRA CONSENSUS DOCUMENT doi:10.1093/europace/eux240 Antithrombotic therapy in atrial fibrillation associated with valvular heart disease: a joint consensus document from the European Heart Rhythm Association (EHRA) and European Society of Cardiology Working Group on Thrombosis, endorsed by the ESC Working Group on Valvular Heart Disease, Cardiac Arrhythmia Society of Southern Africa (CASSA), Heart Rhythm Society (HRS), Asia Pacific Heart Rhythm Society (APHRS), South African Heart (SA Heart) Association and Sociedad Latinoamericana de Estimulacion Cardıaca y Electrofisiologıa (SOLEACE) Gregory Y. H. Lip1*, Jean Philippe Collet2, Raffaele de Caterina3, Laurent Fauchier4, Deirdre A. Lane5, Torben B. Larsen6, Francisco Marin7, Joao Morais8, Calambur Narasimhan9, Brian Olshansky10, Luc Pierard11, Tatjana Potpara12, Nizal Sarrafzadegan13, Karen Sliwa14, Gonzalo Varela15, Gemma Vilahur16, Thomas Weiss17, Giuseppe Boriani18 and Bianca Rocca19 Document Reviewers: Bulent Gorenek20 (Reviewer Coordinator), Irina Savelieva21, Christian Sticherling22, Gulmira Kudaiberdieva23, Tze-Fan Chao24, Francesco Violi25, Mohan Nair26, Leandro Zimerman27, Jonathan Piccini28, Robert Storey29, Sigrun Halvorsen30, Diana Gorog31, Andrea Rubboli32, Ashley Chin33 and Robert Scott-Millar34 * Corresponding author. Tel/fax: þ44 121 5075503. E-mail address: [email protected] Published on behalf of the European Society of Cardiology. All rights reserved. VC The Author 2017. For permissions, please email: [email protected]. 2 G.Y.H. Lip 1Institute of Cardiovascular Sciences, University of Birmingham and Aalborg Thrombosis Research Unit, Department of Clinical Medicine, Aalborg University, Denmark (Chair, representing EHRA); 2Sorbonne Universite´ Paris 6, ACTION Study Group, Institut De Cardiologie, Groupe Hoˆpital Pitie´-Salpetrie`re (APHP), INSERM UMRS 1166, Paris, France; 3Institute of Cardiology, ‘G. -
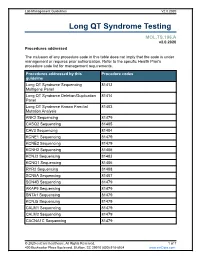
Long QT Syndrome Testing
Lab Management Guidelines V2.0.2020 Long QT Syndrome Testing MOL.TS.196.A v2.0.2020 Procedures addressed The inclusion of any procedure code in this table does not imply that the code is under management or requires prior authorization. Refer to the specific Health Plan's procedure code list for management requirements. Procedures addressed by this Procedure codes guideline Long QT Syndrome Sequencing 81413 Multigene Panel Long QT Syndrome Deletion/Duplication 81414 Panel Long QT Syndrome Known Familial 81403 Mutation Analysis ANK2 Sequencing 81479 CASQ2 Sequencing 81405 CAV3 Sequencing 81404 KCNE1 Sequencing 81479 KCNE2 Sequencing 81479 KCNH2 Sequencing 81406 KCNJ2 Sequencing 81403 KCNQ1 Sequencing 81406 RYR2 Sequencing 81408 SCN5A Sequencing 81407 SCN4B Sequencing 81479 AKAP9 Sequencing 81479 SNTA1 Sequencing 81479 KCNJ5 Sequencing 81479 CALM1 Sequencing 81479 CALM2 Sequencing 81479 CACNA1C Sequencing 81479 © 2020 eviCore healthcare. All Rights Reserved. 1 of 7 400 Buckwalter Place Boulevard, Bluffton, SC 29910 (800) 918-8924 www.eviCore.com Lab Management Guidelines V2.0.2020 What is Long QT syndrome Definition Long QT Syndrome (LQTS) is caused by mutations in a number of genes, most of which are related to the functioning of sodium or potassium ion channels in the heart.1 Testing may offer prognostic information in some cases, as specific genes and even specific mutations within those genes may have some correlation to risk for sudden death, effectiveness of beta-blocker therapy, and preventive strategies.1-4 Signs and symptoms of long QT syndrome (LQTS) are variable, but may include a prolonged QT interval on an electrocardiogram, torsades de pointes, syncope, seizures, cardiac arrest, and sudden cardiac death.1,2 Many patients with LQTS can be largely asymptomatic, with cardiac arrest or sudden cardiac death as the first and only symptom. -

Cardiac Arrest and Ventricular Fibrillation * a Method of Treatment by Electrical Shock
Thorax: first published as 10.1136/thx.7.3.205 on 1 September 1952. Downloaded from Thorax (1952), 7, 205. CARDIAC ARREST AND VENTRICULAR FIBRILLATION * A METHOD OF TREATMENT BY ELECTRICAL SHOCK BY I. K. R. MCMILLANt F. B. COCKETT, AND P. STYLES Froml the Departnment of Cardiology, tile Professorial Surgical Unit, and tile Departalzent of Phlysical Medicine, St. Thomlas's Hospital, London (RECEIVED FOR PUBLICATION APRIL 30, 1952) Cardiac arrest during operation is a subject of VENTRICULAR FIBRILLATION importance to all surgeons. During intra-thoracic The treatment of ventricular fibrillation bv operations cardiac arrest and ventricular fibrilla- electrical shock therapy is not new and was first tion can be differentiated by direct observation of tried experimentally in 1899 (Prevost and Battelli). the heart. In recent years this method has been extensively Ventricular fibrillation is an incoordinated type investigated in the United States and France of contraction which produces no useful beats. (Kouwenhoven and Kay, 1951; Johnson and The typical rippling movements of the ventricular Kirby, 1951 ; Santy and Marion, 1950). muscle are unmistakable when seen or felt with The treatment of ventricular fibrillation by the hand directly on the heart. The treatment of zlectrical shock gives encouraging results in the copyright. a heart that has stopped is cardiac massage and experimental animal, and may be a useful adjunct adequate oxygenation. The treatment of ventri- in the operating theatre during cardiac and general cular fibrillation which we recommend is, first, surgery. Under these conditions shock therapy massage to maintain the oxygen supply to the can be rapidly applied. Provided that adequate heart through the coronary arteries, followed oxygenation is maintained and direct cardiac mas- http://thorax.bmj.com/ rapidly by electrical shocking to restore normal sage promptly initiated and maintained, shock rhythm therapy can be applied without undue haste. -

Effects of Nifekalant Injected Into the Pericardial Space on the Transmural Dispersion of Repolarization in Pig
Showa Univ J Med Sci 19(3), 123 135, September 2007 Original Effects of Nifekalant Injected into the Pericardial Space on the Transmural Dispersion of Repolarization in Pig Hiroyuki ITo, Taku ASANO, Youichi KOBAYASHI, Tatsuya ONUKI, Fumito MwosHI, Taka-aki MATSUYAMA, Yoshino MINOURA, Norikazu WATANABE, Mitsuharu KAWAMURA, Kaoru TANNO and Takashl KATAGIRI Abstract: Transmural dispersion of repolarization (TDR) has been implicated in the onset of ventricular arrhythmia. We investigated the effects of nifekalant (NIF) injected into the pericardial space on TDR and T wave in pig. We injected 50 or 100 mg of NIF into the pericardial space of 11 pigs, and measured the effective refractory period (ERP) between the endocardial and epicardial myocardial cells, as well as QT time and QT peak-end as an index of TDR and T waveforms, respectively. TDR decreased from 56•} 10 msec to 44•}8 msec (P < 0.01), 5.1 min after injection of 100 mg of NIF, although the QTc did not change. At a later time, QTc increased from 457 •} 44 msec before injection to 540•}49 msec (P < 0.01) and TDR recovered to the control level. When 50 mg of NIF was injected, the T wave amplitude decreased from 0.433 •}0.301 mV before injection to 0.107•}0.192 mV (P < 0.01) at 10 min after injection ; 100 mg of NIF caused the T wave amplitude to decrease more rapidly, reaching a negative value at 4.5 min after injection. Injecting NIF into the pericardial space altered ERP, QT, and T waveform, and also decreased TDR. -

The Refractory VF Arrest Patient: a Review of the Current Treatment
1/24/2018 The Refractory VF Arrest Patient: A Review of the Current Treatment Options Marc Conterato, MD, FACEP Office of the Medical Director North Memorial Health Ambulance Service DISCLOSURE STATEMENT • Board Member, MN Resuscitation Consortium - Images of any commercial devices or medications are for illustration purposes only. The inclusion of such images in this presentation does not imply endorsement of any specific device or company. 2 Objectives • Delineate Refractory Ventricular Fibrillation (RVF) • Recurrent VF versus Refractory VF • What is “Electrical Storm” • Current Pre-hospital treatments • Additional hospital treatments • Dual/Double Sequential Defibrillation • ECMO/ECLS-A New Hope? 3 1 1/24/2018 A Standard Scenario • 55 YOF collapses at stop light, and her car rolls into the car in front of her. Airbags do not deploy, and bystanders find her slumped over the steering wheel and pulseless. • First responder/bystanders start CPR, and deliver three AED shocks prior to EMS arrival. • On EMS arrival, a fourth shock is delivered, an IO and alternative airway placed. Automated CPR started. • Epinephrine 2 mg (total) and Amiodarone 300 mg given, and the patient remains in VF. • Patient downtime is now ~25 minutes, and repeat evaluation reveals persistent VF. 4 What are your options • Continue CPR for a total of 30 minutes with recurrent defibrillations, additional epinephrine, bicarbonate and Amiodarone? • “Load and Go” to the local hospital with CPR enroute and continuing the resuscitation? • “Load and Go” to the local CCL (Cardiac Cath Lab) hospital with CPR enroute and continuing the resuscitation, with possible PCI with ongoing CPR? • Call HEMS unit for transfer to CCL hospital, but can they continue effective CPR in the helicopter? • “Load and Go” to a ECMO/ECLS center with CPR enroute and continuing the resuscitation, on the basis they can accept the patient and continue resuscitation? 5 Defining the problem: What is recurrent versus refractory VF (RVF)? • Recurrent VF is a rhythm that terminates with cardioversion, but then recurs rapidly. -

Drugs and Life-Threatening Ventricular Arrhythmia Risk: Results from the DARE Study Cohort
Open Access Research BMJ Open: first published as 10.1136/bmjopen-2017-016627 on 16 October 2017. Downloaded from Drugs and life-threatening ventricular arrhythmia risk: results from the DARE study cohort Abigail L Coughtrie,1,2 Elijah R Behr,3,4 Deborah Layton,1,2 Vanessa Marshall,1 A John Camm,3,4,5 Saad A W Shakir1,2 To cite: Coughtrie AL, Behr ER, ABSTRACT Strengths and limitations of this study Layton D, et al. Drugs and Objectives To establish a unique sample of proarrhythmia life-threatening ventricular cases, determine the characteristics of cases and estimate ► The Drug-induced Arrhythmia Risk Evaluation study arrhythmia risk: results from the the contribution of individual drugs to the incidence of DARE study cohort. BMJ Open has allowed the development of a cohort of cases of proarrhythmia within these cases. 2017;7:e016627. doi:10.1136/ proarrhythmia. Setting Suspected proarrhythmia cases were referred bmjopen-2017-016627 ► These cases have provided crucial safety by cardiologists across England between 2003 and 2011. information, as well as underlying clinical and ► Prepublication history for Information on demography, symptoms, prior medical and genetic data. this paper is available online. drug histories and data from hospital notes were collected. ► Only patients who did not die as a result of the To view these files please visit Participants Two expert cardiologists reviewed data the journal online (http:// dx. doi. proarrhythmia could be included. for 293 referred cases: 130 were included. Inclusion org/ 10. 1136/ bmjopen- 2017- ► Referral of cases by cardiologists alone may have criteria were new onset or exacerbation of pre-existing 016627). -

Atrial Fibrillation (ATRIA) Study
European Journal of Human Genetics (2014) 22, 297–306 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg REVIEW Atrial fibrillation: the role of common and rare genetic variants Morten S Olesen*,1,2,4, Morten W Nielsen1,2,4, Stig Haunsø1,2,3 and Jesper H Svendsen1,2,3 Atrial fibrillation (AF) is the most common cardiac arrhythmia affecting 1–2% of the general population. A number of studies have demonstrated that AF, and in particular lone AF, has a substantial genetic component. Monogenic mutations in lone and familial AF, although rare, have been recognized for many years. Presently, mutations in 25 genes have been associated with AF. However, the complexity of monogenic AF is illustrated by the recent finding that both gain- and loss-of-function mutations in the same gene can cause AF. Genome-wide association studies (GWAS) have indicated that common single-nucleotide polymorphisms (SNPs) have a role in the development of AF. Following the first GWAS discovering the association between PITX2 and AF, several new GWAS reports have identified SNPs associated with susceptibility of AF. To date, nine SNPs have been associated with AF. The exact biological pathways involving these SNPs and the development of AF are now starting to be elucidated. Since the first GWAS, the number of papers concerning the genetic basis of AF has increased drastically and the majority of these papers are for the first time included in a review. In this review, we discuss the genetic basis of AF and the role of both common and rare genetic variants in the susceptibility of developing AF. -

Basic Rhythm Recognition
Electrocardiographic Interpretation Basic Rhythm Recognition William Brady, MD Department of Emergency Medicine Cardiac Rhythms Anatomy of a Rhythm Strip A Review of the Electrical System Intrinsic Pacemakers Cells These cells have property known as “Automaticity”— means they can spontaneously depolarize. Sinus Node Primary pacemaker Fires at a rate of 60-100 bpm AV Junction Fires at a rate of 40-60 bpm Ventricular (Purkinje Fibers) Less than 40 bpm What’s Normal P Wave Atrial Depolarization PR Interval (Normal 0.12-0.20) Beginning of the P to onset of QRS QRS Ventricular Depolarization QRS Interval (Normal <0.10) Period (or length of time) it takes for the ventricles to depolarize The Key to Success… …A systematic approach! Rate Rhythm P Waves PR Interval P and QRS Correlation QRS Rate Pacemaker A rather ill patient……… Very apparent inferolateral STEMI……with less apparent complete heart block RATE . Fast vs Slow . QRS Width Narrow QRS Wide QRS Narrow QRS Wide QRS Tachycardia Tachycardia Bradycardia Bradycardia Regular Irregular Regular Irregular Sinus Brady Idioventricular A-Fib / Flutter Bradycardia w/ BBB Sinus Tach A-Fib VT PVT Junctional 2 AVB / II PSVT A-Flutter SVT aberrant A-Fib 1 AVB 3 AVB A-Flutter MAT 2 AVB / I or II PAT PAT 3 AVB ST PAC / PVC Stability Hypotension / hypoperfusion Altered mental status Chest pain – Coronary ischemic Dyspnea – Pulmonary edema Sinus Rhythm Sinus Rhythm P Wave PR Interval QRS Rate Rhythm Pacemaker Comment . Before . Constant, . Rate 60-100 . Regular . SA Node Upright in each QRS regular . Interval =/< leads I, II, . Look . Interval .12- .10 & III alike .20 Conduction Image reference: Cardionetics/ http://www.cardionetics.com/docs/healthcr/ecg/arrhy/0100_bd.htm Sinus Pause A delay of activation within the atria for a period between 1.7 and 3 seconds A palpitation is likely to be felt by the patient as the sinus beat following the pause may be a heavy beat. -

Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives?
The Science Journal of the Lander College of Arts and Sciences Volume 11 Number 1 Fall 2017 - 2017 Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives? Menachem Braun Touro College Follow this and additional works at: https://touroscholar.touro.edu/sjlcas Part of the Cardiovascular Diseases Commons, and the Medical Genetics Commons Recommended Citation Braun, M. (2017). Should Genetic Testing Be Recommended for Long QT Syndrome Patients and Their Relatives?. The Science Journal of the Lander College of Arts and Sciences, 11(1). Retrieved from https://touroscholar.touro.edu/sjlcas/vol11/iss1/8 This Article is brought to you for free and open access by the Lander College of Arts and Sciences at Touro Scholar. It has been accepted for inclusion in The Science Journal of the Lander College of Arts and Sciences by an authorized editor of Touro Scholar. For more information, please contact [email protected]. Should Genetic Testing be Recommended for Long QT Syndrome Patients and Their Relatives? Menachem Braun Menachem Braun graduated with a BS in Biology in September 2017. Abstract 7KH/RQJ476\QGURPH /476 LVDIDPLOLDOSRWHQWLDOO\IDWDOFDUGLDFDUUK\WKPLD7UDGLWLRQDOO\LWKDVEHHQGLDJQRVHGE\(&* Molecular studies have provided evidence that LQTS can be caused by a range of underlying molecular abnormalities. Genetic research has proven that different forms of LQTS have different genotypic bases. Therefore, it has become possible to diagnose WKHVSHFLÀFW\SHRIGLVHDVHJHQHWLFDOO\7KLVVWXG\H[DPLQHVWKHDGYDQFHPHQWVPDGHLQWKHSDVWWKLUW\\HDUVLQXQGHUVWDQGLQJ /476DQGUHVHDUFKUHJDUGLQJWKHXVHRIJHQHWLFWHVWLQJLQRUGHUWRGHWHUPLQHWKHEHQHÀWVRIJHQHWLFWHVWLQJIRUWKLVGLVHDVH$ survey of original studies which produced the information is presented here, and provides the reader with an understanding of the PHFKDQLFVRIWKHGLVHDVHDQGKRZWKH\GLIIHULQWKHVHYHUDOJHQHWLFYDULDQWV5HVHDUFKVKRZVWKDWWKHEHQHÀWRIJHQHWLFWHVWLQJ must be weighed against the personal implications in may have for a particular patient and his or her family. -

Latest Diagnostic and Treatment Strategies for the Congenital Long QT Syndromes
Latest Diagnostic and Treatment Strategies for the Congenital Long QT Syndromes Michael J. Ackerman, MD, PhD Windland Smith Rice Cardiovascular Genomics Research Professor Professor of Medicine, Pediatrics, and Pharmacology Director, Long QT Syndrome Clinic and the Mayo Clinic Windland Smith Rice Sudden Death Genomics Laboratory President, Sudden Arrhythmia Death Syndromes (SADS) Foundation Learning Objectives to Disclose: • To RECOGNIZE the “faces” (phenotypes) of the congenital long QT syndromes (LQTS) • To CRITIQUE the various diagnostic modalities used in the evaluation of LQTS and UNDERSTAND their limitations • To ASSESS the currently available treatment options for the various LQT syndromes and EVALUATE their efficacy WINDLAND Smith Rice Sudden Death Genomics Laboratory Conflicts of Interest to Disclose: • Consultant – Boston Scientific, Gilead Sciences, Medtronic, St. Jude Medical, and Transgenomic/FAMILION • Royalties – Transgenomic/FAMILION Congenital Long QT Syndrome Normal QT interval QT QT Prolonged QT 1. Syncope 2. Seizures 3. Sudden death Torsades de pointes Congenital Long QT Syndrome Normal QT interval QT QT ♥ 1957 – first clinical description – JLNS ♥ 1960s – RomanoProlonged-Ward QT syndrome ♥ 1983 – “Schwartz/Moss score”1. Syncope ♥ 1991 – first LQTS chromosome locus 2. Seizures ♥ March 10, 1995 – birth of cardiac 3. Sudden channelopathies death Torsades de pointes Congenital Long QT Syndrome Normal QT interval QT QT Prolonged QT 1. Syncope 2. Seizures 3. Sudden death Torsades de pointes Congenital Long QT Syndrome Normal