In Vivo Characterization of Combination Antitumor Chemotherapy with Calcium Channel Blockers and Ci5-Diamminedichloroplatinum(II)1
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Cancer Drug Pharmacology Table
CANCER DRUG PHARMACOLOGY TABLE Cytotoxic Chemotherapy Drugs are classified according to the BC Cancer Drug Manual Monographs, unless otherwise specified (see asterisks). Subclassifications are in brackets where applicable. Alkylating Agents have reactive groups (usually alkyl) that attach to Antimetabolites are structural analogues of naturally occurring molecules DNA or RNA, leading to interruption in synthesis of DNA, RNA, or required for DNA and RNA synthesis. When substituted for the natural body proteins. substances, they disrupt DNA and RNA synthesis. bendamustine (nitrogen mustard) azacitidine (pyrimidine analogue) busulfan (alkyl sulfonate) capecitabine (pyrimidine analogue) carboplatin (platinum) cladribine (adenosine analogue) carmustine (nitrosurea) cytarabine (pyrimidine analogue) chlorambucil (nitrogen mustard) fludarabine (purine analogue) cisplatin (platinum) fluorouracil (pyrimidine analogue) cyclophosphamide (nitrogen mustard) gemcitabine (pyrimidine analogue) dacarbazine (triazine) mercaptopurine (purine analogue) estramustine (nitrogen mustard with 17-beta-estradiol) methotrexate (folate analogue) hydroxyurea pralatrexate (folate analogue) ifosfamide (nitrogen mustard) pemetrexed (folate analogue) lomustine (nitrosurea) pentostatin (purine analogue) mechlorethamine (nitrogen mustard) raltitrexed (folate analogue) melphalan (nitrogen mustard) thioguanine (purine analogue) oxaliplatin (platinum) trifluridine-tipiracil (pyrimidine analogue/thymidine phosphorylase procarbazine (triazine) inhibitor) -

The Immunosuppressive Effects of Long-Term Combination Chemotherapy in Children with Acute Leukemia Inremission1
[CANCER RESEARCH 31, 420-426, April 1971] The Immunosuppressive Effects of Long-Term Combination Chemotherapy in Children with Acute Leukemia in Remission1 Luis Borella and Robert G. Webster Laboratories of Virology and Immunology, St. Jude Children 'sResearch Hospital, Memphis, Tennessee 38101 SUMMARY effects of prolonged maintained combination chemotherapy upon immunocompetence have not been investigated. The immunosuppressive effects of maintenance Knowledge of the immunosuppressive effects of long-term combination chemotherapy given for periods ranging from 8 combination chemotherapy is now urgently needed because of to 28 months to 20 children with acute lymphocytic leukemia the increasing number of prolonged remissions and potential in remission were investigated. There was depression of both 5-year cures among children with acute lymphocytic leukemia the primary antibody production to the hemagglutinin antigen receiving this form of treatment (19). of the Hong Kong influenza virus and the anamnestic response This study was aimed at determining the to the neuraminidase of the same virus. The primary response immunosuppressive effects of long-term combination (hemagglutination inhibition) was affected to a greater extent chemotherapy in children with acute lymphocytic leukemia. than the secondary (neuraminidase inhibition) response. A Several unique features of this investigation were as follows. preferential depression of 2-mercaptoethanol-resistant (a) All patients were in remission and received the same hemagglutination inhibition antibodies (IgG) was also combination chemotherapy continuously for periods ranging observed. One-fourth of all acute lymphocytic leukemia from 8 to 28 months, (b) Patients and controls were patients had low serum IgG levels. In vitro transformation of immunized with the Hong Kong influenza virus vaccine prior lymphocytes was a poor index of immunocompetence. -

Mtorc1/2 Inhibition Preserves Ovarian Function and Fertility During Genotoxic Chemotherapy
mTORC1/2 inhibition preserves ovarian function and fertility during genotoxic chemotherapy Kara N. Goldmana, Devon Chenetteb, Rezina Arjub, Francesca E. Duncanc, David L. Keefea, Jamie A. Grifoa, and Robert J. Schneiderb,d,1 aDivision of Reproductive Endocrinology and Infertility, Department of Obstetrics and Gynecology, New York University School of Medicine, New York,NY 10016; bDepartment of Microbiology, New York University School of Medicine, New York, NY 10016; cDepartment of Obstetrics and Gynecology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611; and dPerlmutter Cancer Center, New York University School of Medicine, New York, NY 10016 Edited by Nahum Sonenberg, McGill University, Montreal, QC, Canada, and approved February 8, 2017 (received for review October 17, 2016) The ovary contains oocytes within immature (primordial) follicles pathway, leading to primordial follicle activation and follicular that are fixed in number at birth. Activation of follicles within this “burnout” (6, 7). fixed pool causes an irreversible decline in reproductive capacity, Ovarian folliculogenesis initiates from the primordial follicle known as the ovarian reserve, until menopause. Premenopausal stage, where an oocyte arrested in prophase of meiosis I and women undergoing commonly used genotoxic (DNA-damaging) surrounded by a single layer of squamous granulosa cells is ac- chemotherapy experience an accelerated loss of the ovarian tivated to grow and transition to a primary follicle, secondary reserve, leading to subfertility and infertility. Therefore, there is follicle, and then ultimately a preovulatory antral follicle (Fig. considerable interest but little effective progress in preserving S1). Most oocytes within the ovary exist in a quiescent state ovarian function during chemotherapy. Here we show that block- within primordial follicles, relatively resistant to antimitotic and ing the kinase mammalian/mechanistic target of rapamycin genotoxic agents (8, 9). -

Introduction to Chemotherapy
2/21/2017 Principles of Chemotherapy EUGENE R. PRZESPOLEWSKI, PHARM.D. BCOP THE JONAH CENTER FOR ONCOLOGY AND HEMATOLOGY ERIE COUNTY MEDICAL CENTER Biology 101 2 Cancer is a complex disease caused by genetic and epigenetic mutations Simply, it is only unregulated cell division “Traditional” chemotherapy highjacks mechanisms of mitosis Understanding chemotherapy needs understanding of Biology 101* * Of course it gets complicated Chemotherapy 3 Merriam-Webster: Chemotherapy: noun: che・mo・ther・a・py Medical: The use of chemical agents in the treatment or control of disease (such as cancer) or mental illness Word originated around 1910 by Paul Ehrlich Developed the first treatment for syphilis, antiserum for diphtheria (Nobel prize in 1908) He also developed the concept of “magic bullet” In the world of pharmacology chemotherapy can be used to treat: Infectious disease Cancer 1 2/21/2017 History of Chemotherapy Begins… 4 World War II 5 Nitrogen Mustards were taboo and not used in battle, however Ready to be used (feared Hitler would use when he was pushed) Bomb raid on Bari, Italy on December 2nd, 1943 Sailors exposed had depletion of bone marrow stores and lymph nodes Goodman and Gilman at Yale discovered murine models with lymphomas responded to nitrogen mustard therapy Convinced a surgeon to treat a single NHL patient with a nitrogen mustard Original trial done in 1943, but data kept secret until 1946 The Lesson of the 1940s 6 Nitrogen Mustards: Alkylation of guanine nucleotides in DNA causing inhibition of cell division -
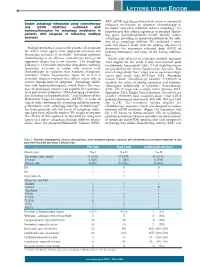
Double Autophagy Stimulation Using Chemotherapy and Mtor Inhibition Combined with Hydroxychloroquine for Autophagy Modulation In
LETTERS TO THE EDITOR AKT- mTOR signaling pathway both serves as a potential Double autophagy stimulation using chemotherapy resistance mechanism to cytotoxic chemotherapy in and mTOR inhibition combined with myeloma10 and when inhibited, induces autophagy,11 we hydroxychloroquine for autophagy modulation in hypothesized that adding rapamycin to standard alkylat- patients with relapsed or refractory multiple ing agent cyclophosphamide would ‘doubly’ induce myeloma autophagy, providing an improved platform for the addi- tion of an autophagy inhibitor. We conducted a safety pilot and phase I study with the primary objective to Multiple myeloma is an incurable plasma cell neoplasm determine the maximum tolerated dose (MTD) of for which novel agents have improved outcomes but hydroxychloroquine and safety of the 4-drug combina- therapeutic resistance is inevitable. Infusional cytotoxic tion. chemotherapy is an effective cytoreductive strategy for Adults with relapsed or refractory multiple myeloma aggressive relapse but is not curative.1 The autophagy were eligible for the study if they had received prior pathway is a lysosome-dependent degradative pathway lenalidomide, bortezomib (Table 1). Full eligibility criteria frequently activated in tumor cells treated with are provided in the Online Supplementary Appendix. This chemotherapy or radiation that mediates therapeutic was a 2-stage study: the 1st stage was an open label single resistance2 (Online Supplementary Figure S1) as it is a center pilot study (July 2011-June 2012; Abramson reversible adaptive response that allows cancer cells to Cancer Center, clinicaltrials.gov identifier: 01396200) to survive therapy-induced apoptosis.3 Autophagy inhibi- establish the safety of adding rapamycin and hydroxy- tion with hydroxychloroquine, which blocks the clear- chloroquine individually to backbone chemotherapy ance of autophagic vesicles,4 can augment the cytotoxici- (n=6). -

Mtor Inhibition Sensitizes Gastric Cancer to Alkylating Chemotherapy in Vivo
ANTICANCER RESEARCH 28 : 3801-3808 (2008) mTOR Inhibition Sensitizes Gastric Cancer to Alkylating Chemotherapy In Vivo DANIEL CEJKA 1, MATTHIAS PREUSSER 2, THORSTEN FUEREDER 1, WOLFGANG SIEGHART 1, JOHANNES WERZOWA 1, SABINE STROMMER 1 and VOLKER WACHECK 1 1Section of Experimental Oncology/Molecular Pharmacology, Department of Clinical Pharmacology, and 2Department of Internal Medicine I, Medical University Vienna, Waehringer Guertel 18-20, 1090 Vienna, Austria Abstract. Background: Gastric cancer is a highly high dose of cyclophosphamide shows synergistic antitumor chemoresistant tumor. Previous studies suggest that cancer activity against gastric cancer in vivo. In potential future cells can be sensitized to standard chemotherapy, and clinical trials, the toxicity of cyclophosphamide in combination especially alkylating agents, by inhibition of mammalian target regimens with everolimus deserves careful evaluation. of rapamycin (mTOR) signaling. The work presented here shows that the mTOR inhibitor everolimus, in combination The mammalian target of rapamycin (mTOR) pathway has with cyclophosphamide, exhibit s synergistic antitumor activity become a major focus of preclinical and clinical cancer in gastric cancer xenografts. Materials and Methods: research (1). Rapamycin inhibits the kinase activity of mTOR, Treatment with everolimus at the minimal effective dose was which has been shown to result in G1 arrest, apoptosis or studied in combination with cyclophosphamide at maximum autophagy, depending on cell type studied (2-4). In gastric tolerated dose in a human gastric cancer severe combined cancer, mTOR activity and components of the mTOR immunodeficient (SCID) mouse xenograft model. Besides signaling network including PTEN, 4E-BP1 and eIF-4 are tumor size, biomarker expression for proliferation (Ki-67), deregulated and correlate with progression of disease, hypoxia (HIF-1α), apoptosis (activated caspase 3), metastasis, and inferior survival (5-9). -

Oral Chemotherapy Drug List
Oral Chemotherapy Drug List (Lista de medicinas para quimioterapia oral) Current (corriente) 10/1/21 Brand versions may not be covered when generics are available, Please check your medication guide (Es posible que las versiones de marca no estén cubiertas cuando hay genéricos disponibles. Consulte su guía de medicamentos) Afinitor imatinib Soltamox Afinitor Disperz Imbruvica Sprycel Alecensa Inlyta Stivarga Alkeran (melphalan) Inqovi Sutent Alunbrig Inrebic Tabloid (thioguanine) anastrozole Iressa Tabrecta Ayvakit Jakafi Tafinlar Balversa Kisqali Tagrisso Bosulif Koselugo Talzenna Braftovi Lenvima tamoxifen Brukinsa leucovorin calcium Tarceva Cabometyx Leukeran Targretin (bexarotene) Calquence Lonsurf Tasigna capecitabine Lorbrena Tazverik Caprelsa Lumakras Temodar (temozolomide) Casodex (bicalutamide) Lynparza Tepmetko CEENU (lomustine) Lysodren Thalomid Cometriq Matulane Tibsovo Copiktra Megace (megesterol) Tretinoin Cotellic Mekinist Trexall Cytoxan (cyclophosphamide) Mektovi Truseltiq Daurismo mercaptopurine Tukysa Droxia (hydroxyurea) methotrexate oral Turalio Emcyt Myleran Tykerb Erivedge Nerlynx Ukoniq Erleada Nexavar Venclexta exemestane Nilandron (nilutamide) Verzenio Eulexin (flutamide) Ninlaro Vitrakvi Evista (raloxifene) Nubeqa Vizimpro Fareston Odomzo Votrient Farydak Onureg Xalkori Femara (letrozole) Orgovyx Xospata Fotivda Pemazyre Xpovio Gavreto Piqray Xtandi Gilotrif Pomalyst Yonsa Gleostine Purixan Zejula Hexalen Qinlock Zelboraf Hycamtin Retevmo Zolinza Hydrea (hydroxyurea) Revlimid Zortress Ibrance Rozlytrek Zydelig Iclusig Rubraca Zykadia Idhifa Rydapt Zytiga Generics = lower case Brands = CAPITAL LETTERS (Genéricos = letras minúscula Marca = LETRAS MAYÚSCULA) Florida Blue is an Independent Licensee of the Blue Cross and Blue Shield Association Page 1 of 1 . -

Chemotherapy and You National Cancer Institute
Support for People With Cancer Chemotherapy and You National Cancer Institute U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES National Institutes of Health Important Phone Numbers Emergency ____________________________________________ Clinic ________________________________________________ Nurse ________________________________________________ Doctor _______________________________________________ Other ________________________________________________ _____________________________________________________ For More Information This is only one of many free books for people with cancer. Here are some others you may find useful: ■■ Biological Therapy ■■ Radiation Therapy and You: Support for People With Cancer ■■ Eating Hints: Before, During, and After Cancer Treatment ■■ Taking Part in Cancer Treatment Research Studies ■■ Thinking About Complementary & Alternative Medicine: A Guide for People With Cancer ■■ Pain Control: A Guide for People With Cancer ■■ When Cancer Returns ■■ Taking Time: Support for People with Cancer These books are available from NCI (the National Cancer Institute). NCI is a federal agency that is part of the National Institutes of Health. Call 1-800-4-CANCER (1-800-422-6237) or visit http://www.cancer.gov. (See page 59 for more information.) *For information about your specific type of cancer, see the PDQ® database. You can also find the database at http://www.cancer.gov. Product or brand names that appear in this book are for example only. The U.S. Government does not endorse any specific product or brand. If products or brands are not mentioned, it does not mean or imply that they are not satisfactory. 1-800-4-CANCER (1-800-422-6237) About This Book Rather than Chemotherapy and You is written for you—someone who is about to read this book receive or is now receiving chemotherapy for cancer. -

For Health Professionals Who Care for Cancer Patients July-August 2008 Website Access At
Volume 11, Number 7 for health professionals who care for cancer patients July-August 2008 Website access at http://www.bccancer.bc.ca/HPI/ChemotherapyProtocols/stupdate.htm I NSIDE THIS ISSUE Benefit List: Changes for Breast Cancer and GUAVPG Revised: BRAVTRNAV, CNCCV, Pediatric Oncology UGICAPIRI, UGICIRB, UGIFFIRB, GIFOLFIRI, Frequently Asked Questions – Steroid Use During GIPGEM, UGIRAJFFOX, UGISORAF, GOCXCRT, Chemotherapy HNAVGEM, LYCHOPR, LYCVPR, LYCYCLO, LYFLUDR, UGUSUNI, LYHDMTXP, LYHDMTXR, Cancer Drug Manual: Complete Revision: ULYMFBEX, ULYRICE, LYRITUX, ULYRMTN, Procarbazine MYPAM, SAAVA Highlights of Changes in Protocols and Pre-Printed Continuing Education – BC Cancer Agency Annual Orders – Irinotecan Protocols, Rituximab Protocols Cancer Conference 2008 and Pre-Printed Orders Editor’s Note – July-August issue Temsirolimus Medication Preparation: Safety Alert Website Resources List of New and Revised Protocols, Pre-Printed Orders and Patient Handouts: New: BRAVGEM, IN TOUCH phone list is provided if additional information is needed. BENEFIT DRUG LIST The following indications have been added to the Benefit Drug List as class II drugs: Gemcitabine palliative therapy for metastatic breast cancer (BRAVGEM) Irinotecan high risk renal tumors in pediatric patients (COG protocol AREN0321) Irinotecan high risk rhabdomyosarcoma in pediatric patients (COG protocol ARSTO431) FREQUENTLY ASKED QUESTIONS: CORTICOSTEROID USE DURING CHEMOTHERAPY The following are some of the frequently asked questions by healthcare professionals around the province on the use of corticosteroids in patients receiving chemotherapy. 1. Does being put on dexamethasone or prednisone during chemotherapy pose any risks to patients? Would this depend on the dose or duration of the corticosteroid? Corticosteroids such as dexamethasone and prednisone may be used for different purposes in patients with cancer. -

Non‐Oncologic Indications for Chemotherapy
7/17/2017 FSHP 2017 ANNUAL MEETING Disclosure #FSHP2017 The speaker of this presentation has the following disclosure: Name Company Role Chemotherapy Outside the Box: Rebecca Gonzalez, Pharm.D., BCOP None N/A Non‐Oncologic Indications For Off label use disclosure: Chemotherapy • This session will include a discussion of off-label treatment and investigational agents not approved by the FDA for use in the US Rebecca Gonzalez, Pharm.D., BCOP Clinical Pharmacist Blood and Marrow Transplantation Moffitt Cancer Center 2017 ANNUAL MEETING #FSHP2017 #FSHP2017 Pharmacist Objectives Technician Objectives 1. Identify non-oncologic indications for specific 1. Identify chemotherapy and biotherapy agents that chemotherapy and biotherapy agents may be used for non-oncologic indications 2. Discuss barriers to distribution and administration of 2. Discuss barriers to distribution of chemotherapy in non- chemotherapy/biotherapy in non-oncology settings oncology settings 3. Review common toxicities and monitoring associated 3. Describe safe storage, preparation, and disposal of with use of specific chemotherapy/biotherapy agents chemotherapy agents used in non-oncology settings 2017 ANNUAL MEETING 2017 ANNUAL MEETING #FSHP2017 #FSHP2017 Background Background: IMIDs • Approximately 80 different diseases result from the Historic treatment New treatment immune system attack on its own cells, tissue and organs • Loss of regulation and differentiation of immune cells • Steroids • Chemotherapy • Irregular function and production cytokines • Production of autoantibodies • Analgesics • Biotherapeutic agents • AD/IMIDs • Non-steroidal anti- Immunosuppressant or inflammatory agents immunomodulating effects • Up to 24 million Americans suffer from AD • Improve symptoms • Cancer: 9 million • Achieve remission • Heart Disease: 22 million AD=autoimmune Disorders; IMIDs=immune-mediated chronic inflammatory diseases IMIDs, immune-mediated chronic inflammatory diseases 2017 ANNUAL MEETING https://www.niaid.nih.gov/diseases-conditions/autoimmune-diseases. -

Table 1: Drug-Drug Interactions of Common Cardiac Drugs and Chemotherapeutic Agents*
Table 1: Drug-Drug Interactions of Common Cardiac Drugs and Chemotherapeutic Agents* Cardiac Drug(s) Enzyme/ Chemotherapy Effect of Drug- Suggested Oncologist Suggested Cardiologist Action Drug† Drug Management Management Interaction Beta-Blockers All beta- Additive Ceritinib Additive Avoid using the combination of ceritinib with beta- blockers clinical bradycardia blockers. If concomitant use is necessary and symptomatic effect bradycardia occurs, hold ceritinib, adjust or discontinue the beta-blocker, and upon recovery resume ceritinib at a reduced dose with frequent monitoring of heart rate.‡ Crizotinib Monitor blood pressure and heart rate regularly. Dose reduction or discontinuation of one of the agents may be necessary if clinically significant bradycardia occurs.‡ Carvedilol P-gp Afatinib ↑ Monitor for adverse Consider alternative agent if inhibition chemotherapy effects of afatinib. If possible. (moderate) drug not well-tolerated, concentration decrease afatinib daily dose by 10 mg. Doxorubicin Monitor for adverse Consider alternative agent if Nilotinib effects of possible. If carvedilol is used for Paclitaxel chemotherapy drug if prevention of anthracycline Pazopanib concomitant therapy is cardiotoxicity, individual risk vs. Vincristine necessary. benefit must be considered. If Vinblastine concomitant therapy is necessary and drug-drug interaction involves QT- prolonging chemotherapy drug, ensure appropriate electrocardiographic (ECG) and electrolyte monitoring. Carvedilol; CYP2D6 Imatinib ↑ beta-blocker Monitor blood pressure -

Chemotherapy – What It Is, How It Helps
Chemotherapy What It Is, How It Helps What’s in this guide? This booklet will explain chemotherapy. Chemotherapy (chemo) is one of the most common treatments for cancer. Chemo may be used alone or with other treatments. If your treatment plan includes chemotherapy, knowing how it works and what to expect can help you make good decisions as you prepare for treatment. If you have more questions, ask your cancer care team to help you. It’s always best to be open and honest with them. That way, they can help you decide which treatment is best for you. Questions about chemotherapy What is chemotherapy? The word “chemotherapy” is what drugs that treat cancer are often called. But, not all drugs used to treat cancer work the same way. Be sure you know what kind of drugs are in your treatment plan. If your treatment plan includes traditional or standard chemotherapy, knowing how it works and what to expect can help you make good decisions as you prepare for treatment. How is chemo used to treat cancer? Many different kinds of chemo drugs are used to treat cancer – either alone or with other drugs or treatments. The drugs are different in what they are made of, how they are given, how strong they are, and what side effects they have. Your doctor figures out the best treatment options to offer based on the kind of cancer you have and how much cancer is in your body (this is called the cancer’s stage). Your cancer care team will talk to you about the goals of chemo before you start treatment.