Target Enrichment of Thousands of Ultraconserved Elements Sheds New Light on Early Relationships Within New World Sparrows (Aves: Passerellidae)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The First Central American Record of White-Throated Sparrow Zonotrichia Albicollis
Cotinga 26 The first Central American record of White-throated Sparrow Zonotrichia albicollis Oliver Komar,Adri de Gelder†, Kerem A. Boyla, George Angehr, Philip Balderamos and Gary Allport Received 22 August 2005; final revision accepted 22 December 2005 Cotinga 26(2006): 43–45 Reportamos un vagabundo de Gorrión Gorjiblanco Zonotrichia albicollis en el cayo Half Moon, a 73 km al sureste de la Ciudad de Belice, siendo el primer registro de esta especie norteamericana para Belice y para América Central. El ave apareció en la isla pequeña al mismo tiempo que se encontraban varias aves transeuntes que migraban hacía el norte. Especulamos que el gorrión hizo una parada durante su migración después de invernar más al sur. On 18 May 2005, we visited tiny Half Moon Caye, darker grey breast and cheeks. Other field marks at the edge of Lighthouse Reef, 73 km south-east of clearly noted were the streaked brown back, brown Belize City, Belize, in the western Caribbean. The wings with two indistinct wingbars, long brown, island covers just c.18 ha; it has sandy wooded slightly notched tail, pale grey rear underparts, thickets on the western four-fifths, and open, buffy flanks and pink legs. It occasionally gave a coconut Cocos nucifera-shaded beach on the eastern characteristic very high tseeet. end. The caye harbours a nesting colony of Red- The sparrow often sought the shade of young footed Booby Sula sula and Magnificent coconut palms or piles of dried palm fronds. Frigatebird Fregata magnificens. Although it picked frequently amongst the sand, we At 07h45, near the Belize Audubon reserve were unable to see if the bird found food, although headquarters on the north-facing beach at the east some seeds from the sparse beach grass and other end of the island (17°12’N 87°32’W), AdG found a plants, as well as small ants and other insects, largish sparrow on drying coconut fronds between probably were in the sand. -

L O U I S I a N A
L O U I S I A N A SPARROWS L O U I S I A N A SPARROWS Written by Bill Fontenot and Richard DeMay Photography by Greg Lavaty and Richard DeMay Designed and Illustrated by Diane K. Baker What is a Sparrow? Generally, sparrows are characterized as New World sparrows belong to the bird small, gray or brown-streaked, conical-billed family Emberizidae. Here in North America, birds that live on or near the ground. The sparrows are divided into 13 genera, which also cryptic blend of gray, white, black, and brown includes the towhees (genus Pipilo), longspurs hues which comprise a typical sparrow’s color (genus Calcarius), juncos (genus Junco), and pattern is the result of tens of thousands of Lark Bunting (genus Calamospiza) – all of sparrow generations living in grassland and which are technically sparrows. Emberizidae is brushland habitats. The triangular or cone- a large family, containing well over 300 species shaped bills inherent to most all sparrow species are perfectly adapted for a life of granivory – of crushing and husking seeds. “Of Louisiana’s 33 recorded sparrows, Sparrows possess well-developed claws on their toes, the evolutionary result of so much time spent on the ground, scratching for seeds only seven species breed here...” through leaf litter and other duff. Additionally, worldwide, 50 of which occur in the United most species incorporate a substantial amount States on a regular basis, and 33 of which have of insect, spider, snail, and other invertebrate been recorded for Louisiana. food items into their diets, especially during Of Louisiana’s 33 recorded sparrows, Opposite page: Bachman Sparrow the spring and summer months. -

A Comprehensive Multilocus Assessment of Sparrow (Aves: Passerellidae) Relationships ⇑ John Klicka A, , F
Molecular Phylogenetics and Evolution 77 (2014) 177–182 Contents lists available at ScienceDirect Molecular Phylogenetics and Evolution journal homepage: www.elsevier.com/locate/ympev Short Communication A comprehensive multilocus assessment of sparrow (Aves: Passerellidae) relationships ⇑ John Klicka a, , F. Keith Barker b,c, Kevin J. Burns d, Scott M. Lanyon b, Irby J. Lovette e, Jaime A. Chaves f,g, Robert W. Bryson Jr. a a Department of Biology and Burke Museum of Natural History and Culture, University of Washington, Box 353010, Seattle, WA 98195-3010, USA b Department of Ecology, Evolution, and Behavior, University of Minnesota, 100 Ecology Building, 1987 Upper Buford Circle, St. Paul, MN 55108, USA c Bell Museum of Natural History, University of Minnesota, 100 Ecology Building, 1987 Upper Buford Circle, St. Paul, MN 55108, USA d Department of Biology, San Diego State University, San Diego, CA 92182, USA e Fuller Evolutionary Biology Program, Cornell Lab of Ornithology, Cornell University, 159 Sapsucker Woods Road, Ithaca, NY 14950, USA f Department of Biology, University of Miami, 1301 Memorial Drive, Coral Gables, FL 33146, USA g Universidad San Francisco de Quito, USFQ, Colegio de Ciencias Biológicas y Ambientales, y Extensión Galápagos, Campus Cumbayá, Casilla Postal 17-1200-841, Quito, Ecuador article info abstract Article history: The New World sparrows (Emberizidae) are among the best known of songbird groups and have long- Received 6 November 2013 been recognized as one of the prominent components of the New World nine-primaried oscine assem- Revised 16 April 2014 blage. Despite receiving much attention from taxonomists over the years, and only recently using molec- Accepted 21 April 2014 ular methods, was a ‘‘core’’ sparrow clade established allowing the reconstruction of a phylogenetic Available online 30 April 2014 hypothesis that includes the full sampling of sparrow species diversity. -

Sonoran Joint Venture Bird Conservation Plan Version 1.0
Sonoran Joint Venture Bird Conservation Plan Version 1.0 Sonoran Joint Venture 738 N. 5th Avenue, Suite 102 Tucson, AZ 85705 520-882-0047 (phone) 520-882-0037 (fax) www.sonoranjv.org May 2006 Sonoran Joint Venture Bird Conservation Plan Version 1.0 ____________________________________________________________________________________________ Acknowledgments We would like to thank all of the members of the Sonoran Joint Venture Technical Committee for their steadfast work at meetings and for reviews of this document. The following Technical Committee meetings were devoted in part or total to working on the Bird Conservation Plan: Tucson, June 11-12, 2004; Guaymas, October 19-20, 2004; Tucson, January 26-27, 2005; El Palmito, June 2-3, 2005, and Tucson, October 27-29, 2005. Another major contribution to the planning process was the completion of the first round of the northwest Mexico Species Assessment Process on May 10-14, 2004. Without the data contributed and generated by those participants we would not have been able to successfully assess and prioritize all bird species in the SJV area. Writing the Conservation Plan was truly a group effort of many people representing a variety of agencies, NGOs, and universities. Primary contributors are recognized at the beginning of each regional chapter in which they participated. The following agencies and organizations were involved in the plan: Arizona Game and Fish Department, Audubon Arizona, Centro de Investigación Cientifica y de Educación Superior de Ensenada (CICESE), Centro de Investigación de Alimentación y Desarrollo (CIAD), Comisión Nacional de Áreas Naturales Protegidas (CONANP), Instituto del Medio Ambiente y el Desarrollo (IMADES), PRBO Conservation Science, Pronatura Noroeste, Proyecto Corredor Colibrí, Secretaría de Medio Ambiente y Recursos Naturales (SEMARNAT), Sonoran Institute, The Hummingbird Monitoring Network, Tucson Audubon Society, U.S. -

Ultimate Bolivia Tour Report 2019
Titicaca Flightless Grebe. Swimming in what exactly? Not the reed-fringed azure lake, that’s for sure (Eustace Barnes) BOLIVIA 8 – 29 SEPTEMBER / 4 OCTOBER 2019 LEADER: EUSTACE BARNES Bolivia, indeed, THE land of parrots as no other, but Cotingas as well and an astonishing variety of those much-loved subfusc and generally elusive denizens of complex uneven surfaces. Over 700 on this tour now! 1 BirdQuest Tour Report: Ultimate Bolivia 2019 www.birdquest-tours.com Blue-throated Macaws hoping we would clear off and leave them alone (Eustace Barnes) Hopefully, now we hear of colourful endemic macaws, raucous prolific birdlife and innumerable elusive endemic denizens of verdant bromeliad festooned cloud-forests, vast expanses of rainforest, endless marshlands and Chaco woodlands, each ringing to the chorus of a diverse endemic avifauna instead of bleak, freezing landscapes occupied by impoverished unhappy peasants. 2 BirdQuest Tour Report: Ultimate Bolivia 2019 www.birdquest-tours.com That is the flowery prose, but Bolivia IS that great destination. The tour is no longer a series of endless dusty journeys punctuated with miserable truck-stop hotels where you are presented with greasy deep-fried chicken and a sticky pile of glutinous rice every day. The roads are generally good, the hotels are either good or at least characterful (in a good way) and the food rather better than you might find in the UK. The latter perhaps not saying very much. Palkachupe Cotinga in the early morning light brooding young near Apolo (Eustace Barnes). That said, Bolivia has work to do too, as its association with that hapless loser, Che Guevara, corruption, dust and drug smuggling still leaves the country struggling to sell itself. -

Ecuador: HARPY EAGLE & EAST ANDEAN FOOTHILLS EXTENSION
Tropical Birding Trip Report Ecuador: HARPY EAGLE & East Andean Foothills Extension (Jan-Feb 2021) A Tropical Birding custom extension Ecuador: HARPY EAGLE & EAST ANDEAN FOOTHILLS EXTENSION th nd 27 January - 2 February 2021 The main motivation for this custom extension was this Harpy Eagle. This was one of an unusually accessible nesting pair near the Amazonian town of Limoncocha that provided a worthy add-on to The Andes Introtour in northwest Ecuador that preceded this (Jose Illanes/Tropical Birding Tours). Guided by Jose Illanes Birds in the photos within this report are denoted in RED, all photos were taken by the Tropical Birding guide. 1 www.tropicalbirding.com +1-409-515-9110 [email protected] Tropical Birding Trip Report Ecuador: HARPY EAGLE & East Andean Foothills Extension (Jan-Feb 2021) INTRODUCTION This custom extension trip was set up for one person who simply could not get enough of Ecuador…John had just finished Ecuador: The Andes Introtour, in the northwest of the country, and also joined the High Andes Extension to that tour, which sampled the eastern highlands too. However, he was still missing vast chunks of this small country that is bursting with bird diversity. Most importantly, he was keen to get in on the latest “mega bird” in Ecuador, a very accessible Harpy Eagle nest, near a small Amazonian town, which had been hitting the local headlines and drawing the few birding tourists in the country at this time to come see it. With this in mind, TROPICAL BIRDING has been offering custom add-ons to all of our Ecuador offerings (for 2021 and 2022) to see this Harpy Eagle pair, with only three extra days needed to see it. -

21 Sep 2018 Lists of Victims and Hosts of the Parasitic
version: 21 Sep 2018 Lists of victims and hosts of the parasitic cowbirds (Molothrus). Peter E. Lowther, Field Museum Brood parasitism is an awkward term to describe an interaction between two species in which, as in predator-prey relationships, one species gains at the expense of the other. Brood parasites "prey" upon parental care. Victimized species usually have reduced breeding success, partly because of the additional cost of caring for alien eggs and young, and partly because of the behavior of brood parasites (both adults and young) which may directly and adversely affect the survival of the victim's own eggs or young. About 1% of all bird species, among 7 families, are brood parasites. The 5 species of brood parasitic “cowbirds” are currently all treated as members of the genus Molothrus. Host selection is an active process. Not all species co-occurring with brood parasites are equally likely to be selected nor are they of equal quality as hosts. Rather, to varying degrees, brood parasites are specialized for certain categories of hosts. Brood parasites may rely on a single host species to rear their young or may distribute their eggs among many species, seemingly without regard to any characteristics of potential hosts. Lists of species are not the best means to describe interactions between a brood parasitic species and its hosts. Such lists do not necessarily reflect the taxonomy used by the brood parasites themselves nor do they accurately reflect the complex interactions within bird communities (see Ortega 1998: 183-184). Host lists do, however, offer some insight into the process of host selection and do emphasize the wide variety of features than can impact on host selection. -

Henslow's Sparrow
Table of Contents I. Introduction………………………………………………………… 1 II. Species Account……………………………………………………. 1 A. Taxonomy Description…………………………………………. 1 1. Original Description…………………………………….. 1 2. Taxonomic Description…………………………………. 3 B. Historical and Current Distribution……………………………... 3 1. Description of Habitats and Locations of Occurrence….. 3 2. Known Collection Sites………………………………… 5 3. Associated Species and Communities………………...... 5 C. Population Sizes and Abundance………………………………. 7 D. Reproduction…………………………………………………… 8 E. Food and Feeding Requirements……………………………...... 8 F. Other Pertinent Information and Summary…………………...... 9 III. Ownership of Properties…………………………………………… 9 IV. Potential Threats…………………………………………………… 10 V. Protective Laws…………………………………………………...... 12 A. Federal………………………………………………………...... 12 B. State…………………………………………………………...... 12 VI. Recovery………………………………………………………….... 12 A. Objectives………………………………………………………. 12 B. Recovery Criteria……………………………………………..... 12 VII. Narrative Outline…………………………………………………... 13 1. Additional Species Information Needs…………………………. 13 2. Management Activities for Maintaining Species Populations and for Species Recovery………………………………………....... 14 VIII. Costs of Recovery Plan Implementation…………………………… 15 Table 1 Henslow’s sparrow densities for sites of occurrence………………. 18 Figure 1……………………………………………………………………………..... 20 Literature Cited………………………………………………………………………. 21 i Acknowledgements We would like to thank all of the individuals who assisted with or provided information for the completion of this plan. Most -

Bird) Species List
Aves (Bird) Species List Higher Classification1 Kingdom: Animalia, Phyllum: Chordata, Class: Reptilia, Diapsida, Archosauria, Aves Order (O:) and Family (F:) English Name2 Scientific Name3 O: Tinamiformes (Tinamous) F: Tinamidae (Tinamous) Great Tinamou Tinamus major Highland Tinamou Nothocercus bonapartei O: Galliformes (Turkeys, Pheasants & Quail) F: Cracidae Black Guan Chamaepetes unicolor (Chachalacas, Guans & Curassows) Gray-headed Chachalaca Ortalis cinereiceps F: Odontophoridae (New World Quail) Black-breasted Wood-quail Odontophorus leucolaemus Buffy-crowned Wood-Partridge Dendrortyx leucophrys Marbled Wood-Quail Odontophorus gujanensis Spotted Wood-Quail Odontophorus guttatus O: Suliformes (Cormorants) F: Fregatidae (Frigatebirds) Magnificent Frigatebird Fregata magnificens O: Pelecaniformes (Pelicans, Tropicbirds & Allies) F: Ardeidae (Herons, Egrets & Bitterns) Cattle Egret Bubulcus ibis O: Charadriiformes (Sandpipers & Allies) F: Scolopacidae (Sandpipers) Spotted Sandpiper Actitis macularius O: Gruiformes (Cranes & Allies) F: Rallidae (Rails) Gray-Cowled Wood-Rail Aramides cajaneus O: Accipitriformes (Diurnal Birds of Prey) F: Cathartidae (Vultures & Condors) Black Vulture Coragyps atratus Turkey Vulture Cathartes aura F: Pandionidae (Osprey) Osprey Pandion haliaetus F: Accipitridae (Hawks, Eagles & Kites) Barred Hawk Morphnarchus princeps Broad-winged Hawk Buteo platypterus Double-toothed Kite Harpagus bidentatus Gray-headed Kite Leptodon cayanensis Northern Harrier Circus cyaneus Ornate Hawk-Eagle Spizaetus ornatus Red-tailed -

Updating the Distribution of the Sierra Madre Sparrow Xenospiza Baileyi Across Central Mexico: Historical Records, New Localities, and Conservation Perspectives
VOLUME 15, ISSUE 1, ARTICLE 15 Ortega-Álvarez, R., R. Calderón-Parra, U. Martínez Molina, F. Martínez Molina, G. Martínez Molina, Y. Martínez Molina, A. Martínez Villagrán, J. Martínez Freire, R. Vásquez Robles, D. García Loaeza, J. Martínez García, S. García Loaeza, N. I. Garduño López, and L. A. Sánchez-González. 2020. Updating the distribution of the Sierra Madre Sparrow Xenospiza baileyi across central Mexico: historical records, new localities, and conservation perspectives. Avian Conservation and Ecology 15(1):15. https://doi.org/10.5751/ACE-01573-150115 Copyright © 2020 by the author(s). Published here under license by the Resilience Alliance. Research Paper Updating the distribution of the Sierra Madre Sparrow Xenospiza baileyi across central Mexico: historical records, new localities, and conservation perspectives Rubén Ortega-Álvarez 1, Rafael Calderón-Parra 2, Ulises Martínez Molina 2, Fredy Martínez Molina 2, Gabriel Martínez Molina 2, Yuridia Martínez Molina 2, Agustín Martínez Villagrán 2, Josué Martínez Freire 2, Rocío Vásquez Robles 2, Delfino García Loaeza 2, Jaciel Martínez García 2, Sarai García Loaeza 2, Nancy Isabel Garduño López 2 and Luis A. Sánchez-González 1 1Museo de Zoología "Alfonso L. Herrera," Departamento de Biología Evolutiva, Facultad de Ciencias, Universidad Nacional Autónoma de México, Ciudad de México, México, 2Brigada de Monitoreo Biológico Milpa Alta, San Pablo Oztotepec, Milpa Alta, Ciudad de México, México ABSTRACT. The Sierra Madre Sparrow (Xenospiza baileyi) is an endangered species microendemic to subalpine grasslands of central and northwestern Mexico. Given that land use change is highly dynamic in central Mexico, there is a need for a continuous monitoring of the distribution and conservation status of the species across the region. -

FIELD GUIDES BIRDING TOURS: Colombia: Bogota, the Magdalena
Field Guides Tour Report Colombia: Bogota, the Magdalena Valley, and Santa Marta 2014 Jan 11, 2014 to Jan 27, 2014 Jesse Fagan & Trevor Ellery For our tour description, itinerary, past triplists, dates, fees, and more, please VISIT OUR TOUR PAGE. A fun group and the most productive tour we have had to date! We observed 582 bird taxa in 17 days of birding, which beat our record last year of 555 by a bunch. As we fine-tune our birding route and learn more about Colombian birds things just seem to get better and better. This year we saw 33 endemics and loads of interesting subspecies and near-endemics. Highlights included a female Blue- billed Curassow, Kelp Gull(s) at Los Camerones (only the second time it has been recorded in Colombia), Dwarf and Pavonine cuckoos (the latter a lifer for Trevor!), a splendid Crested Owl, Sapphire- bellied Hummingbird (nice comparisons with Sapphire-throated), Double-banded Graytail in the coffee finca below Reinita Cielo Azul lodge, the always elusive Santa Marta Bush-Tyrant and antpitta, Turquoise Dacnis, and singing Yellow-bellied Siskin. It is really hard to pick just one from so many! I want to thank all of you again for a really enjoyable trip. Thanks also to Trevor Ellery, our local guide, and Giovanni, our driver, for their hard work. I look forward to seeing you again in the field. Bird On. --Jesse a.k.a. Motmot (from Lima, Peru) KEYS FOR THIS LIST One of the following keys may be shown in brackets for individual species as appropriate: * = heard only, I = introduced, E = endemic, N = nesting, a = austral migrant, b = boreal migrant This dazzling Black-cheeked Mountain-Tanager is a Santa Marta endemic; it was one of 33 endemics we tallied on this species-rich tour. -
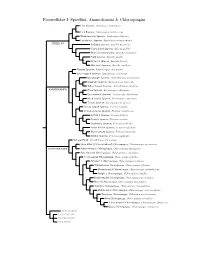
Passerellidae Species Tree
Passerellidae I: Spizellini, Ammodramini & Chlorospingini Lark Sparrow, Chondestes grammacus Lark Bunting, Calamospiza melanocorys Black-throated Sparrow, Amphispiza bilineata Five-striped Sparrow, Amphispiza quinquestriata SPIZELLINI Chipping Sparrow, Spizella passerina Clay-colored Sparrow, Spizella pallida Black-chinned Sparrow, Spizella atrogularis Field Sparrow, Spizella pusilla Brewer’s Sparrow, Spizella breweri Worthen’s Sparrow, Spizella wortheni Tumbes Sparrow, Rhynchospiza stolzmanni Stripe-capped Sparrow, Rhynchospiza strigiceps Grasshopper Sparrow, Ammodramus savannarum Grassland Sparrow, Ammodramus humeralis Yellow-browed Sparrow, Ammodramus aurifrons AMMODRAMINI Olive Sparrow, Arremonops rufivirgatus Green-backed Sparrow, Arremonops chloronotus Black-striped Sparrow, Arremonops conirostris Tocuyo Sparrow, Arremonops tocuyensis Rufous-winged Sparrow, Peucaea carpalis Cinnamon-tailed Sparrow, Peucaea sumichrasti Botteri’s Sparrow, Peucaea botterii Cassin’s Sparrow, Peucaea cassinii Bachman’s Sparrow, Peucaea aestivalis Stripe-headed Sparrow, Peucaea ruficauda Black-chested Sparrow, Peucaea humeralis Bridled Sparrow, Peucaea mystacalis Tanager Finch, Oreothraupis arremonops Short-billed (Yellow-whiskered) Chlorospingus, Chlorospingus parvirostris CHLOROSPINGINI Yellow-throated Chlorospingus, Chlorospingus flavigularis Ashy-throated Chlorospingus, Chlorospingus canigularis Sooty-capped Chlorospingus, Chlorospingus pileatus Wetmore’s Chlorospingus, Chlorospingus wetmorei White-fronted Chlorospingus, Chlorospingus albifrons Brown-headed