1 ORIGINAL ARTICLE Role of SCN5A Coding and Non-Coding
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Next Generation Sequencing for Molecular Confirmation of Hereditary
Arch Cardiol Mex. 2015;85(1):68---72 www.elsevier.com.mx SPECIAL ARTICLE Next generation sequencing for molecular confirmation of hereditary sudden cardiac death syndromes a,∗ b b b Manlio F. Márquez , David Cruz-Robles , Selene Ines-Real , Gilberto Vargas-Alarcón , a Manuel Cárdenas a Departamento de Electrofisiología, Instituto Nacional de Cardiología Ignacio Chávez, México, D.F., Mexico b Departamento de Biología Molecular, Instituto Nacional de Cardiología Ignacio Chávez, México, D.F., Mexico Received 26 March 2014; accepted 8 December 2014 KEYWORDS Abstract Hereditary sudden cardiac death syndromes comprise a wide range of diseases result- Arrhythmias; ing from alteration in cardiac ion channels. Genes involved in these syndromes represent diverse Hereditary sudden mutations that cause the altered encoding of the diverse proteins constituting these channels, cardiac death thus affecting directly the currents of the corresponding ions. In the present article we will syndromes; briefly review how to arrive to a clinical diagnosis and we will present the results of molecular Right ventricle genetic studies made in Mexican subjects attending the SCD Syndromes Clinic of the National arrhythmogenic Institute of Cardiology of Mexico City. cardiomyopathy; © 2014 Instituto Nacional de Cardiología Ignacio Chávez. Published by Masson Doyma México Brugada syndrome S.A. All rights reserved. PALABRAS CLAVE Confirmación diagnóstica molecular mediante secuenciación masiva de nueva Arritmias; generación (‘‘next generation sequencing’’) en síndromes hereditarios de muerte Síndromes súbita cardíaca hereditarios de Resumen Los síndromes hereditarios de muerte súbita cardíaca comprenden una amplia gama muerte súbita; Displasia de enfermedades resultantes de la alteración en los canales iónicos cardíacos. Los genes implicados en estos síndromes presentan mutaciones que causan alteraciones de las diversas Arritmogénica del proteínas que constituyen estos canales y que, por lo tanto, afectan directamente a las difer- ventriculo derecho; entes corrientes iónicas. -

Non-Coding Rnas in the Cardiac Action Potential and Their Impact on Arrhythmogenic Cardiac Diseases
Review Non-Coding RNAs in the Cardiac Action Potential and Their Impact on Arrhythmogenic Cardiac Diseases Estefania Lozano-Velasco 1,2 , Amelia Aranega 1,2 and Diego Franco 1,2,* 1 Cardiovascular Development Group, Department of Experimental Biology, University of Jaén, 23071 Jaén, Spain; [email protected] (E.L.-V.); [email protected] (A.A.) 2 Fundación Medina, 18016 Granada, Spain * Correspondence: [email protected] Abstract: Cardiac arrhythmias are prevalent among humans across all age ranges, affecting millions of people worldwide. While cardiac arrhythmias vary widely in their clinical presentation, they possess shared complex electrophysiologic properties at cellular level that have not been fully studied. Over the last decade, our current understanding of the functional roles of non-coding RNAs have progressively increased. microRNAs represent the most studied type of small ncRNAs and it has been demonstrated that miRNAs play essential roles in multiple biological contexts, including normal development and diseases. In this review, we provide a comprehensive analysis of the functional contribution of non-coding RNAs, primarily microRNAs, to the normal configuration of the cardiac action potential, as well as their association to distinct types of arrhythmogenic cardiac diseases. Keywords: cardiac arrhythmia; microRNAs; lncRNAs; cardiac action potential Citation: Lozano-Velasco, E.; Aranega, A.; Franco, D. Non-Coding RNAs in the Cardiac Action Potential 1. The Electrical Components of the Adult Heart and Their Impact on Arrhythmogenic The adult heart is a four-chambered organ that propels oxygenated blood to the entire Cardiac Diseases. Hearts 2021, 2, body. It is composed of atrial and ventricular chambers, each of them with distinct left and 307–330. -

Identification of Key Genes and Pathways Involved in Response To
Deng et al. Biol Res (2018) 51:25 https://doi.org/10.1186/s40659-018-0174-7 Biological Research RESEARCH ARTICLE Open Access Identifcation of key genes and pathways involved in response to pain in goat and sheep by transcriptome sequencing Xiuling Deng1,2†, Dong Wang3†, Shenyuan Wang1, Haisheng Wang2 and Huanmin Zhou1* Abstract Purpose: This aim of this study was to investigate the key genes and pathways involved in the response to pain in goat and sheep by transcriptome sequencing. Methods: Chronic pain was induced with the injection of the complete Freund’s adjuvant (CFA) in sheep and goats. The animals were divided into four groups: CFA-treated sheep, control sheep, CFA-treated goat, and control goat groups (n 3 in each group). The dorsal root ganglions of these animals were isolated and used for the construction of a cDNA= library and transcriptome sequencing. Diferentially expressed genes (DEGs) were identifed in CFA-induced sheep and goats and gene ontology (GO) enrichment analysis was performed. Results: In total, 1748 and 2441 DEGs were identifed in CFA-treated goat and sheep, respectively. The DEGs identi- fed in CFA-treated goats, such as C-C motif chemokine ligand 27 (CCL27), glutamate receptor 2 (GRIA2), and sodium voltage-gated channel alpha subunit 3 (SCN3A), were mainly enriched in GO functions associated with N-methyl- D-aspartate (NMDA) receptor, infammatory response, and immune response. The DEGs identifed in CFA-treated sheep, such as gamma-aminobutyric acid (GABA)-related DEGs (gamma-aminobutyric acid type A receptor gamma 3 subunit [GABRG3], GABRB2, and GABRB1), SCN9A, and transient receptor potential cation channel subfamily V member 1 (TRPV1), were mainly enriched in GO functions related to neuroactive ligand-receptor interaction, NMDA receptor, and defense response. -

Cardiogenetics Testing Reference Guide December 2018
Cardiogenetics Testing reference guide December 2018 Why Choose Ambry More than 1 in 200 people have an inherited cardiovascular condition. Ambry’s mission is to provide the most advanced genetic testing information available to help you identity those at-risk and determine the best treatment options. If we know a patient has a disease-causing genetic change, not only does it mean better disease management, it also indicates that we can test others in the family and provide them with potentially life-saving information. Diseases and Testing Options cardiomyopathies arrhythmias Hypertrophic Cardiomyopathy (HCMNext) Catecholaminergic Polymorphic Ventricular Dilated Cardiomyopathy (DCMNext) Tachycardia (CPVTNext) Arrhythmogenic Right Ventricular Long QT Syndrome, Short QT Syndrome, Cardiomyopathy (ARVCNext) Brugada Syndrome (LongQTNext, RhythmNext) Cardiomyopathies (CMNext, CardioNext) Arrhythmias (RhythmNext, CardioNext) other cardio conditions Transthyretin Amyloidosis (TTR) familial hypercholesterolemia Noonan Syndrome (NoonanNext) and lipid disorders Hereditary Hemorrhagic Telangiectasia Familial Hypercholesterolemia (FHNext) (HHTNext) Sitosterolemia (Sitosterolemia Panel) Comprehensive Lipid Menu thoracic aortic aneurysms (CustomNext-Cardio) and dissections Familial Chylomicronemia Syndrome (FCSNext) Thoracic Aneurysms and Dissections, aortopathies (TAADNext) Marfan Syndrome (TAADNext) Ehlers-Danlos Syndrome (TAADNext) Targeted Panels Gene Comparison ALL PANELS HAVE A TURNAROUND TIME OF 2-3 WEEKS arrhythmias CPVTNext CPVTNext CASQ2, -

Singular Localization of Sodium Channel &Beta
ARTICLE Received 2 May 2014 | Accepted 9 Oct 2014 | Published 21 Nov 2014 DOI: 10.1038/ncomms6525 Singular localization of sodium channel b4 subunit in unmyelinated fibres and its role in the striatum Haruko Miyazaki1,2,3,4, Fumitaka Oyama2,5, Ritsuko Inoue6, Toshihiko Aosaki6, Takaya Abe7, Hiroshi Kiyonari7, Yoshihiro Kino1,2,3,4, Masaru Kurosawa1,2,3,4, Jun Shimizu8, Ikuo Ogiwara9, Kazuhiro Yamakawa9, Yoshinori Koshimizu10, Fumino Fujiyama4,10,11, Takeshi Kaneko10, Hideaki Shimizu12, Katsuhiro Nagatomo13, Katsuya Yamada13, Tomomi Shimogori3, Nobutaka Hattori14, Masami Miura6 & Nobuyuki Nukina1,2,3,4 Voltage-gated Na þ channel b-subunits are multifunctional molecules that modulate Na þ channel activity and regulate cell adhesion, migration and neurite outgrowth. b-subunits including b4 are known to be highly concentrated in the nodes of Ranvier and axon initial segments in myelinated axons. Here we show diffuse b4 localization in striatal projection fibres using transgenic mice that express fluorescent protein in those fibres. These axons are unmyelinated, forming large, inhibitory fibre bundles. Furthermore, we report b4 dimer expression in the mouse brain, with high levels of b4 dimers in the striatal projection fascicles, suggesting a specific role of b4 in those fibres. Scn4b-deficient mice show a resurgent Na þ current reduction, decreased repetitive firing frequency in medium spiny neurons and increased failure rates of inhibitory postsynaptic currents evoked with repetitive stimulation, indicating an in vivo channel regulatory role of b4 in the striatum. 1 Department of Neuroscience for Neurodegenerative Disorders, Juntendo University Graduate School of Medicine, 2-1-1 Hongo, Bunkyo-ku, Tokyo 113-8421, Japan. -

Ion Channels 3 1
r r r Cell Signalling Biology Michael J. Berridge Module 3 Ion Channels 3 1 Module 3 Ion Channels Synopsis Ion channels have two main signalling functions: either they can generate second messengers or they can function as effectors by responding to such messengers. Their role in signal generation is mainly centred on the Ca2 + signalling pathway, which has a large number of Ca2+ entry channels and internal Ca2+ release channels, both of which contribute to the generation of Ca2 + signals. Ion channels are also important effectors in that they mediate the action of different intracellular signalling pathways. There are a large number of K+ channels and many of these function in different + aspects of cell signalling. The voltage-dependent K (KV) channels regulate membrane potential and + excitability. The inward rectifier K (Kir) channel family has a number of important groups of channels + + such as the G protein-gated inward rectifier K (GIRK) channels and the ATP-sensitive K (KATP) + + channels. The two-pore domain K (K2P) channels are responsible for the large background K current. Some of the actions of Ca2 + are carried out by Ca2+-sensitive K+ channels and Ca2+-sensitive Cl − channels. The latter are members of a large group of chloride channels and transporters with multiple functions. There is a large family of ATP-binding cassette (ABC) transporters some of which have a signalling role in that they extrude signalling components from the cell. One of the ABC transporters is the cystic − − fibrosis transmembrane conductance regulator (CFTR) that conducts anions (Cl and HCO3 )and contributes to the osmotic gradient for the parallel flow of water in various transporting epithelia. -

Transcriptomic Uniqueness and Commonality of the Ion Channels and Transporters in the Four Heart Chambers Sanda Iacobas1, Bogdan Amuzescu2 & Dumitru A
www.nature.com/scientificreports OPEN Transcriptomic uniqueness and commonality of the ion channels and transporters in the four heart chambers Sanda Iacobas1, Bogdan Amuzescu2 & Dumitru A. Iacobas3,4* Myocardium transcriptomes of left and right atria and ventricles from four adult male C57Bl/6j mice were profled with Agilent microarrays to identify the diferences responsible for the distinct functional roles of the four heart chambers. Female mice were not investigated owing to their transcriptome dependence on the estrous cycle phase. Out of the quantifed 16,886 unigenes, 15.76% on the left side and 16.5% on the right side exhibited diferential expression between the atrium and the ventricle, while 5.8% of genes were diferently expressed between the two atria and only 1.2% between the two ventricles. The study revealed also chamber diferences in gene expression control and coordination. We analyzed ion channels and transporters, and genes within the cardiac muscle contraction, oxidative phosphorylation, glycolysis/gluconeogenesis, calcium and adrenergic signaling pathways. Interestingly, while expression of Ank2 oscillates in phase with all 27 quantifed binding partners in the left ventricle, the percentage of in-phase oscillating partners of Ank2 is 15% and 37% in the left and right atria and 74% in the right ventricle. The analysis indicated high interventricular synchrony of the ion channels expressions and the substantially lower synchrony between the two atria and between the atrium and the ventricle from the same side. Starting with crocodilians, the heart pumps the blood through the pulmonary circulation and the systemic cir- culation by the coordinated rhythmic contractions of its upper lef and right atria (LA, RA) and lower lef and right ventricles (LV, RV). -
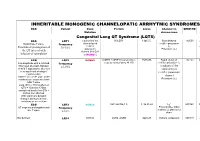
Inheritable Arrhytmia Syndromes
INHERITABLE MONOGENIC CHANNELOPATIC ARRHYTMIC SYNDROMES ECG Variant Gene Protein Locus Channel on OMIM NO IP Mutation chromosome Congenital Long QT Syndrome (LQTS) ECG LQT1 α subunit of the KvLQT1 11p15.5. Slow delayed 192500 AD slow delayed rectifier potassium or Broad base T wave. Frequency rectifier I AR Paradoxical prolongation of 30-35% ks potassium Potassium (IKs) the QT interval with channel (KvLQT1 infusion of epinephrine or KCNQ1) ECG LQT2 KCNH2 (HERG + MiRP1) human ether- 7q35q36 Rapid delayed 152.427 AD a-go-go related gene HERG rectifier potassium I Low-amplitude with a notched, Frequency kr bifurcated alternant, biphasic 25-30% α subunit of the or bifid T appearance due to a rapid delayed very significant slowing of rectifier potassium repolarization. channel KCNH2 on L413P and L559H mutations are associated with Potassium (IKr) bifid T wave Long. QTc > 470ms affected QTc = 450ms to 470ms considered border line QTc < 450ms non affected. With significant dynamic changes during heart rate variations or on exertion + ECG LQT3 SCN5A hH1 and NaV1.5 3, 3p 21-24 INa 600163 AD Prolonged I + influx ST segment prolongation and Frequency Na late T wave. in phase 2, plateau or 5-10% dome Not defined LQT4 KCNJ2 ANK2, ANKB 4q25-27 Sodium, potassium 600919 AR and calcium + Not defined LQT5 KCNE1 minK K Efflux 176261 AD Not defined LQT6 KCNE2 MiRP1 603796 MiRP1 Frequency rare Modest prolongation of QT LQT7 Andersen KCNJ2 Kir2.1 Ik1 Reduction 170390 AD interval, prominent U wave, Great reduction in I Tawil-syndrome k1 frequent PVCs, PVT, result in the bidirectional VT. -

High-Throughput Behavioral Phenotyping in the Expanded Panel of BXD Recombinant Inbred Strains
Genes, Brain and Behavior (2010) 9: 129–159 © 2009 The Authors Journal compilation © 2009 Blackwell Publishing Ltd/International Behavioural and Neural Genetics Society High-throughput behavioral phenotyping in the expanded panel of BXD recombinant inbred strains V. M. Philip†,S.Duvvuru†,B.Gomero†, despair and related neurological phenomena; pain T. A. Ansah‡,C.D.Blaha§,M.N.Cook§, sensitivity; stress sensitivity; anxiety; hyperactivity and K. M. Hamre¶, W. R. Lariviere∗∗, sleep/wake cycles. All traits have been measured in D. B. Matthews††,‡‡,G.Mittleman§, both sexes in approximately 70 strains of the recently D. Goldowitz§§ andE.J.Chesler†,∗ expanded panel of BXD RI strains. Sex differences and heritability estimates were obtained for each trait, and a comparison of early (N = 32) and recent (N = 37) † Systems Genetics Group, Biosciences Division, Oak Ridge BXD RI lines was performed. Primary data are publicly ‡ National Laboratory, Oak Ridge TN, Department of available for heritability, sex difference and genetic Neurobiology and Neurotoxicology, Meharry Medical College, analyses using the MouseTrack database, and are also § Nashville, TN, Department of Psychology, The University of available in GeneNetwork.org for quantitative trait locus ¶ Memphis, Memphis, TN, Departments of Anatomy and (QTL) detection and genetic analysis of gene expression. Neurobiology, University of Tennessee Health Science Center, Together with the results of related studies, these data ** Memphis, TN, Departments of Anesthesiology and form a public resource -

Therapeutic Approaches to Genetic Ion Channelopathies and Perspectives in Drug Discovery
fphar-07-00121 May 7, 2016 Time: 11:45 # 1 REVIEW published: 10 May 2016 doi: 10.3389/fphar.2016.00121 Therapeutic Approaches to Genetic Ion Channelopathies and Perspectives in Drug Discovery Paola Imbrici1*, Antonella Liantonio1, Giulia M. Camerino1, Michela De Bellis1, Claudia Camerino2, Antonietta Mele1, Arcangela Giustino3, Sabata Pierno1, Annamaria De Luca1, Domenico Tricarico1, Jean-Francois Desaphy3 and Diana Conte1 1 Department of Pharmacy – Drug Sciences, University of Bari “Aldo Moro”, Bari, Italy, 2 Department of Basic Medical Sciences, Neurosciences and Sense Organs, University of Bari “Aldo Moro”, Bari, Italy, 3 Department of Biomedical Sciences and Human Oncology, University of Bari “Aldo Moro”, Bari, Italy In the human genome more than 400 genes encode ion channels, which are transmembrane proteins mediating ion fluxes across membranes. Being expressed in all cell types, they are involved in almost all physiological processes, including sense perception, neurotransmission, muscle contraction, secretion, immune response, cell proliferation, and differentiation. Due to the widespread tissue distribution of ion channels and their physiological functions, mutations in genes encoding ion channel subunits, or their interacting proteins, are responsible for inherited ion channelopathies. These diseases can range from common to very rare disorders and their severity can be mild, Edited by: disabling, or life-threatening. In spite of this, ion channels are the primary target of only Maria Cristina D’Adamo, University of Perugia, Italy about 5% of the marketed drugs suggesting their potential in drug discovery. The current Reviewed by: review summarizes the therapeutic management of the principal ion channelopathies Mirko Baruscotti, of central and peripheral nervous system, heart, kidney, bone, skeletal muscle and University of Milano, Italy Adrien Moreau, pancreas, resulting from mutations in calcium, sodium, potassium, and chloride ion Institut Neuromyogene – École channels. -

Graded Co-Expression of Ion Channel, Neurofilament, and Synaptic Genes in Fast- Spiking Vestibular Nucleus Neurons
Research Articles: Cellular/Molecular Graded co-expression of ion channel, neurofilament, and synaptic genes in fast- spiking vestibular nucleus neurons https://doi.org/10.1523/JNEUROSCI.1500-19.2019 Cite as: J. Neurosci 2019; 10.1523/JNEUROSCI.1500-19.2019 Received: 26 June 2019 Revised: 11 October 2019 Accepted: 25 October 2019 This Early Release article has been peer-reviewed and accepted, but has not been through the composition and copyediting processes. The final version may differ slightly in style or formatting and will contain links to any extended data. Alerts: Sign up at www.jneurosci.org/alerts to receive customized email alerts when the fully formatted version of this article is published. Copyright © 2019 the authors 1 Graded co-expression of ion channel, neurofilament, and synaptic genes in fast-spiking 2 vestibular nucleus neurons 3 4 Abbreviated title: A fast-spiking gene module 5 6 Takashi Kodama1, 2, 3, Aryn Gittis, 3, 4, 5, Minyoung Shin2, Keith Kelleher2, 3, Kristine Kolkman3, 4, 7 Lauren McElvain3, 4, Minh Lam1, and Sascha du Lac1, 2, 3 8 9 1 Johns Hopkins University School of Medicine, Baltimore MD, 21205 10 2 Howard Hughes Medical Institute, La Jolla, CA, 92037 11 3 Salk Institute for Biological Studies, La Jolla, CA, 92037 12 4 Neurosciences Graduate Program, University of California San Diego, La Jolla, CA, 92037 13 5 Carnegie Mellon University, Pittsburgh, PA, 15213 14 15 Corresponding Authors: 16 Takashi Kodama ([email protected]) 17 Sascha du Lac ([email protected]) 18 Department of Otolaryngology-Head and Neck Surgery 19 The Johns Hopkins University School of Medicine 20 Ross Research Building 420, 720 Rutland Avenue, Baltimore, Maryland, 21205 21 22 23 Conflict of Interest 24 The authors declare no competing financial interests. -

Cardiovascular Diseases Genetic Testing Program Information
Cardiovascular Diseases Genetic Testing Program Description: Congenital Heart Disease Panels We offer comprehensive gene panels designed to • Congenital Heart Disease Panel (187 genes) diagnose the most common genetic causes of hereditary • Heterotaxy Panel (114 genes) cardiovascular diseases. Testing is available for congenital • RASopathy/Noonan Spectrum Disorders Panel heart malformation, cardiomyopathy, arrythmia, thoracic (31 genes) aortic aneurysm, pulmonary arterial hypertension, Marfan Other Panels syndrome, and RASopathy/Noonan spectrum disorders. • Pulmonary Arterial Hypertension (PAH) Panel Hereditary cardiovascular disease is caused by variants in (20 genes) many different genes, and may be inherited in an autosomal dominant, autosomal recessive, or X-linked manner. Other Indications: than condition-specific panels, we also offer single gene Panels: sequencing for any gene on the panels, targeted variant • Confirmation of genetic diagnosis in a patient with analysis, and targeted deletion/duplication analysis. a clinical diagnosis of cardiovascular disease Tests Offered: • Carrier or pre-symptomatic diagnosis identification Arrythmia Panels in individuals with a family history of cardiovascular • Comprehensive Arrhythmia Panel (81 genes) disease of unknown genetic basis • Atrial Fibrillation (A Fib) Panel (28 genes) Gene Specific Sequencing: • Atrioventricular Block (AV Block) Panel (7 genes) • Confirmation of genetic diagnosis in a patient with • Brugada Syndrome Panel (21 genes) cardiovascular disease and in whom a specific