Single Compartment Micro Direct Glucose Fuel Cell
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Energy Analysis and Fabrication of Photovoltaic Thermal Water Electrolyzer and Ion Transport Through Modified Nanoporous Membranes
ENERGY ANALYSIS AND FABRICATION OF PHOTOVOLTAIC THERMAL WATER ELECTROLYZER AND ION TRANSPORT THROUGH MODIFIED NANOPOROUS MEMBRANES BY MUHAMMED ENES ORUC DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemical Engineering in the Graduate College of the University of Illinois at Urbana-Champaign, 2014 Urbana, Illinois Doctoral Committee: Professor Hong Yang, Chair Professor Ralph G. Nuzzo, Director of Research Professor Paul J.A. Kenis Assistant Professor David W. Flaherty Abstract Hydrogen is an environmentally sustainable energy carrier that can be stored. It is not found naturally and therefore must be artificially produced. We can obtain hydrogen from renewable energy, such solar and wind energy, which is environmentally clean. One such a promising options is via electrolysis using electricity from a photovoltaic generator. In the first part of the dissertation we studied a microfluidic energy conversion device to produce hydrogen. Particularly, we proposed a new integrated system – a so-called “photovoltaic thermal water electrolyzer (PVTE)” – which consists of PV cells positioned on top of a planar micro-water electrolyzers in order to harness waste heat as a storable form of energy. The concept of PVTE has the outputs such as electricity and thermal storage, and also it provides hydrogen production efficiently. First, we provided a comprehensive analysis of the overall efficiency of the PVTE system. COMSOL Multiphysics software was used to predict the temperatures for the electrolyte and the PV cells operating at various temperatures and solar fluxes. Moreover, hourly and monthly efficiency analyses were accomplished for Phoenix, AZ in the year 2010. This new integrated approach is advantageous over conventional PV modules (Chapter 2). -

Catalysis Science & Technology
Catalysis Science & Technology Accepted Manuscript This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/catalysis Page 1 of 29 Catalysis Science & Technology Catalysis Science & Technology RSC Publishing MINIREVIEW Hydrogen Energy Future with Formic Acid: A Cite this: DOI: 10.1039/x0xx00000x Renewable Chemical Hydrogen Storage System ,a ,b ,c Manuscript Received 00th August 2015, Ashish Kumar Singh* , Suryabhan Singh* and Abhinav Kumar* Accepted 00th August 2015 DOI: 10.1039/x0xx00000x Formic acid, the simplest carboxylic acid, is found in nature or can be easily synthesized in laboratory (major by-product of some second generation biorefinery processes), an important www.rsc.org/ chemical due to its myriad applications in pharmaceuticals and industries. -

Curriculum Vitae
CURRICULUM VITA FOR SU HA EDUCATION Ph.D. in Chemical Engineering, University of Illinois, Urbana, IL Graduation: October, 2005 Advisor: Richard Masel Thesis: Direct Formic Acid Fuel Cells For Alternative Portable Power Sources M.S. in Chemical Engineering, University of Illinois, Urbana, IL Graduation: October 2003 Advisor: Richard Masel Thesis: Direct Formic Acid Polymer Electrolyte Membrane Fuel Cell B.S. in Chemical Engineering, North Carolina State University, Raleigh, NC Graduated with University Honors, June 2000; University Scholars Program Undergraduate Research Advisor: Saad Khan Research Project: Rheology of Protein Gels Synthesized Through a Combined Enzymatic and Heat Treatment Method PROFESSIONAL EXPERIENCE Associate Professor 2011-Present School of Chemical Engineering and Bioengineering Washington State University, Pullman, WA Assistant Professor 2005-2011 School of Chemical Engineering and Bioengineering Washington State University, Pullman, WA Graduate Research Assistant 2000-2005 Department of Chemical Engineering University of Illinois, Urbana, IL Undergraduate Research Assistant 1998-2000 Department of Chemical Engineering North Carolina State University, Raleigh, NC HONORS AND AWARDS 1. Outstanding Teaching Award, Chemical Engineering Department, Washington State University (2008). 2. 3rd Place, Dr. Bernard S. Baker Award for Fuel Cell Research, Fuel Cell Seminar and Fuel Cell Energy, Inc. (2005). 3. Nominated for the Glenn Award, ACS National Meeting (2005). 4. The 205th Meeting of the Electrochemical Society Travel Award, Energy Technology Division of the Electrochemical Society (2004). 5. Vodafone-U.S. Foundation Graduate Fellowship, University of Illinois (2003). 6. Finalist, The College Invention Competition 2003, The National Inventors Hall of Fame (2003). 7. Winner and Best of the Best, The 9th Annual Undergraduate Research Symposium, North Carolina State University (2000). -

Preparation of Pt-Pd Catalysts for Direct Formic Acid Fuel Cell and Their Characteristics
Korean J. Chem. Eng., 24(3), 518-521 (2007) SHORT COMMUNICATION Preparation of Pt-Pd catalysts for direct formic acid fuel cell and their characteristics Ki Ho Kim, Jae-Keun Yu*, Hyo Song Lee**, Jae Ho Choi, Soon Young Noh, Soo Kyung Yoon***, Chang-Soo Lee, Taek-Sung Hwang and Young Woo Rhee† Department of Chemical Engineering, Chungnam National University, Daejeon 305-764, Korea *Korea Institute of Footwear and Leather Technology, Busan 614-100, Korea **Korea Environment and Resources Corperation, Incheon 404-170, Korea ***Netpreneur Co., Ltd., Seongnam 463-870, Korea (Received 28 August 2006 • accepted 14 November 2006) Abstract−Pt-Pd catalysts were prepared by using the spontaneous deposition method and their characteristics were analyzed in a direct formic acid fuel cell (DFAFC). Effects of calcination temperature and atmosphere on the cell per- formance were investigated. The calcination temperatures were 300, 400 and 500 oC and the calcination atmospheres were air and nitrogen. The fuel cell with the catalyst calcined at 400 oC showed the best cell performance of 58.8 mW/ cm2. The effect of calcination atmosphere on the overall performance of fuel cell was negligible. The fuel cell with catalyst calcined at air atmosphere showed high open circuit potential (OCP) of 0.812 V. Also the effects of anode and cathode catalyst loadings on the DFAFC performance using Pt-Pd (1 : 1) catalyst were investigated to optimize the catalyst loading. The catalyst loading had a significant effect on the fuel cell performance. Especially, the fuel cell with anode catalyst loading of 4 mg/cm2 and cathode catalyst loading of 5 mg/cm2 showed the best power density of 64.7 mW/ cm2 at current density of 200 mA/cm2. -

Development of Membraneless Mixed-Reactant Microfluidic Fuel Cells: Electrocatalysis and Evolution Through Numerical Simulation
Université du Québec Institut National de la Recherche Scientifique Centre Énergie, Matériaux et Télécommunications DEVELOPMENT OF MEMBRANELESS MIXED-REACTANT MICROFLUIDIC FUEL CELLS: ELECTROCATALYSIS AND EVOLUTION THROUGH NUMERICAL SIMULATION Par Juan Carlos Abrego Martínez Thèse présentée pour l’obtention du grade de Philosophiae Doctor (Ph.D.) en sciences de l’énergie et des matériaux Jury d’évaluation Président du jury et Andreas Ruediger examinateur interne Professeur à l’INRS-ÉMT Examinateur externe Ricardo Izquierdo Professeur à l’École de technologie supérieure (ETS) Examinateur externe Sasha Omanovic Professeur à l’Université de McGill Directeur de recherche Mohamed Mohamedi Professeur à l’INRS-ÉMT Codirecteur de recherche Shuhui Sun Professeur à l’INRS-ÉMT © Droits réservés de Juan Carlos Abrego Martínez, 4 Juin 2020 ACKNOWLEDGMENTS First and foremost, I would like to thank my supervisor, Prof. Mohamed Mohamedi for offering me the opportunity to undertake my PhD studies in his research group, for providing the tools and trusting me to carry out this project and for the excellent guidance and support throughout this period. I also express my gratitude to my co-supervisor, Prof. Shuhui Sun for closely following the research progress and for his valuable contribution for accomplishing my PhD project. I thank the Jury members, Prof. Andreas Ruediger, Prof. Ricardo Izquierdo and Prof. Sasha Omanovic for agreeing and taking the time to review and evaluate this work. I would like to thank my group colleagues, Youling Wang, Alonso Moreno, Haixia Wang, Naser Mohammadi, Xiaoying Zheng, Khawtar Hasan and Soraya Bouachma, for sharing their knowledge and experience through discussions and experiments in the laboratory. -

Co-Laminar Flow Cells for Electrochemical Energy Conversion
Co-laminar flow cells for electrochemical energy conversion by Marc-Antoni Goulet M.Sc. (Physics), McMaster University, 2008 B.Sc., McGill University, 2006 Dissertation Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in the School of Engineering Science Faculty of Applied Sciences Marc-Antoni Goulet 2016 SIMON FRASER UNIVERSITY Summer 2016 Approval Name: Marc-Antoni Goulet Degree: Doctor of Philosophy Title: Co-laminar flow cells for electrochemical energy conversion Examining Committee: Chair: Amr Marzouk Lecturer Erik Kjeang Senior Supervisor Associate Professor Michael Eikerling Supervisor Professor Gary Wang Supervisor Professor Edward Park Internal Examiner Professor Matthew Mench External Examiner Professor Department of Mechanical, Aerospace and Biomedical Engineering University of Tennessee, Knoxville Date Defended/Approved: May 2nd, 2016 ii Abstract A recently developed class of electrochemical cell based on co-laminar flow of reactants through porous electrodes is investigated. New architectures are designed and assessed for fuel recirculation and rechargeable battery operation. Extensive characterization of cells is performed to determine most sources of voltage loss during operation. To this end, a specialized flow cell technique is developed to mitigate mass transport limitations and measure kinetic rates of reaction on flow-through porous electrodes. This technique is used in in conjunction with cyclic voltammetry and electrochemical impedance spectroscopy to evaluate different treatments for enhancing the rates of vanadium redox reactions on carbon paper electrodes. It is determined that surface area enhancements are the most effective way for increasing redox reaction rates and thus a novel in situ flowing deposition method is conceived to achieve this objective at minimal cost. -

The Mechanism of Direct Formic Acid Fuel Cell Using Pd, Pt and Pt-Ru
Extended Summary 本文は pp.721-726 The Mechanism of Direct Formic Acid Fuel Cell Using Pd, Pt and Pt-Ru Nobuyuki Kamiya Non-member (Yokohama National University) Yan Liu Non-member (Yokohama National University) Shigenori Mitsushima Non-member (Yokohama National University) Ken-ichiro Ota Non-member (Yokohama National University) Yasuyuki Tsutsumi Member (Electric Power Development Co., Ltd.) Naoya Ogawa Non-member (Electric Power Development Co., Ltd.) Norihiro Kon Non-member (Ibaraki University) Mika Eguchi Non-member (Ibaraki University) Keywords : formic acid, Pd, 2-propanol, dehydrogenation, fuel cell The electro-oxidation of formic acid, 2-propanol and methanol Slow scan voltammogram (SSV) and chronoamperometry on Pd black, Pd/C, Pt-Ru/C and Pt/C has been investigated to clear measurements showed that the activity of formic acid oxidation the reaction mechanism. It was suggested that the formic acid is increased in the following order: Pd black > Pd 30wt.%/C > dehydrogenated on Pd surface and the hydrogen is occluded in the Pt50wt.%/C > 27wt.%Pt-13wt.%Ru/C. A large oxidation current Pd lattice. Thus obtained hydrogen acts like pure hydrogen for formic acid was found at a low overpotential on the palladium supplied from the outside and the cell performance of the direct electrocatalysts. These results indicate that formic acid is mainly formic acid fuel cell showed as high as that of a hydrogen-oxygen oxidized through a dehydrogenation reaction. For the oxidation of fuel cell. 2-propanol did not show such dehydrogenation reaction 2-propanol and methanol, palladium was not effective, and on Pd catalyst. Platinum and Pt-Ru accelerated the oxidation of 27wt.%Pt-13wt.%Ru/C showed the best oxidation activity. -
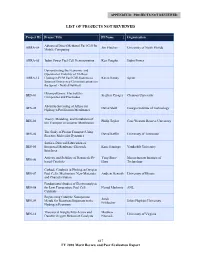
DOE Hydrogen Program 2010 Annual Merit Review and Peer Evaluation
APPENDIX D: PROJECTS NOT REVIEWED LIST OF PROJECTS NOT REVIEWED Project ID Project Title PI Name Organization Advanced Direct Methanol Fuel Cell for ARRA-04 Jim Fletcher University of North Florida Mobile Computing ARRA-05 Jadoo Power Fuel Cell Demonstration Ken Vaughn Jadoo Power Demonstrating the Economic and Operational Viability of 72-Hour ARRA-12 Hydrogen PEM Fuel Cell Systems to Kevin Kenny Sprint Support Emergency Communications on the Sprint - Nextel Network Fluoropolymers, Electrolytes, BES-01 Stephen Creager Clemson University Composites and Electrodes Ab-initio Screening of Alloys for BES-02 David Sholl Georgia Institute of Technology Hydrogen Purification Membranes Theory, Modeling, and Simulation of BES-03 Philip Taylor Case Western Reserve University Ion Transport in Ionomer Membranes The Study of Proton Transport Using BES-04 David Keffer University of Tennessee Reactive Molecular Dynamics Surface-Directed Fabrication of BES-05 Integrated Membrane-Electrode Kane Jennings Vanderbilt University Interfaces Activity and Stability of Nanoscale Pt- Yang Shao- Massachusetts Institute of BES-06 based Catalysts Horn Technology Cathode Catalysis in Hydrogen/Oxygen BES-07 Fuel Cells: Mechanism, New Materials, Andrew Gewirth University of Illinois and Characterization Fundamental Studies of Electrocatalysis BES-08 for Low Temperature Fuel Cell Nenad Markovic ANL Catalysts Engineering Catalytic Nanoporous Jonah BES-09 Metals for Reactions Important to the Johns Hopkins University Erlebacher Hydrogen Economy Theoretical Insights Into -

Membraneless Hydrogen Bromine Laminar Flow Battery for Large
Membraneless Hydrogen Bromine Laminar Flow Battery for Large-Scale Energy Storage by William Allan Braff Submitted to the Department of Mechanical Engineering in partial fulfillment of the requirements for the degree of Doctor of Philosophy at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY February 2014 c Massachusetts Institute of Technology 2014. All rights reserved. Author.............................................................. Department of Mechanical Engineering December 19, 2013 Certified by. Cullen R. Buie Assistant Professor of Mechanical Engineering Thesis Supervisor Certified by. Martin Z. Bazant Professor of Chemical Engineering and Mathematics Thesis Supervisor Accepted by . David E. Hardt Chairman, Department Committee on Graduate Theses 2 Membraneless Hydrogen Bromine Laminar Flow Battery for Large-Scale Energy Storage by William Allan Braff Submitted to the Department of Mechanical Engineering on December 19, 2013, in partial fulfillment of the requirements for the degree of Doctor of Philosophy Abstract Electrochemical energy storage systems have been considered for a range of potential large-scale energy storage applications. These applications vary widely, both in the order of magnitude of energy storage that is required and the rate at which energy must be charged and discharged. One such application aids the integration of renew- able energy technologies onto the electrical grid by shifting the output from renewable energy resources to periods of high demand, relaxing transmission and distribution requirements and reducing the need for fossil fuel burning plants. Although the mar- ket need for such solutions is well known, existing technologies are still too expensive to compete with conventional combustion-based solutions. In this thesis, the hydrogen bromine laminar flow battery (HBFLB) is proposed and examined for its potential to provide low cost energy storage using the rapid reaction kinetics of hydrogen-bromine reaction pairs and a membrane-less laminar flow battery architecture. -

(12) United States Patent (10) Patent No.: US 7,618,725 B2 Masel Et Al
US007618725B2 (12) United States Patent (10) Patent No.: US 7,618,725 B2 Masel et al. (45) Date of Patent: Nov. 17, 2009 (54) LOW CONTAMINANT FORMIC ACID FUEL 3,983,010 A * 9, 1976 Rauch et al. .................. 203/15 FOR DIRECT LIQUID FUEL CELL 4,126,748 A 11, 1978 Scholz 4,262,140 A 4, 1981. Bott (75) Inventors: Richard I. Masel, Champaign, IL (US); 5,294,740 A * 3/1994 Kiefer et al. ................ 562/609 5,393,922 A 2, 1995 Sen Yimin Zhu, Urbana, IL (US); Zakia 5,599.979 A 2/1997 Berg Kahn, Palatine, IL (US); Malcolm Man, 5,633,402 A 5/1997 Berg Vancouver (CA) 5,763,662 A 6/1998 Ikariya 5,869,739 A 2/1999 Ikariya (73) Assignee: The Board of Trustees of the 5,879,915 A 3, 1999 Loubiere University of Illinois, Urbana, IL (US) 5,904,740 A * 5/1999 Davis .......................... 44,385 2003/0050506 A1 3/2003 Auer et al. (*) Notice: Subject to any disclaimer, the term of this 2003/0198852 A1 10, 2003 Masel et al. .................. 429/30 patent is extended or adjusted under 35 U.S.C. 154(b) by 550 days. OTHER PUBLICATIONS Yiminet al., High power density direct formic acid fuel cells, Journal (21) Appl. No.: 11/079,893 of Power Sources 130 (2004) 8-14.* (22) Filed: Mar. 14, 2005 (Continued) (65) Prior Publication Data Primary Examiner Patrick Ryan Assistant Examiner Thomas H. Parsons US 2006/OO597.69 A1 Mar. 23, 2006 (74) Attorney, Agent, or Firm Greer, Burns & Crain, Ltd. -

Benchmarking Catalysts for Formic Acid/Formate Electrooxidation
molecules Review Benchmarking Catalysts for Formic Acid/Formate Electrooxidation Scott J. Folkman 1,* , Jesús González-Cobos 2,*, Stefano Giancola 1,†, Irene Sánchez-Molina 1 and José Ramón Galán-Mascarós 1,3 1 Institute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology (BIST), Av. Paisos Catalans, 16, 43007 Tarragona, Spain; [email protected] (S.G.); [email protected] (I.S.-M.); [email protected] (J.R.G.-M.) 2 Institut de Recherches sur la Catalyse et l’Environnement de Lyon, UMR 5256, CNRS, Université Claude Bernard Lyon 1, 2 Avenue A. Einstein, 69626 Villeurbanne, France 3 ICREA, Pg. Llu’ıs Companys 23, 08010 Barcelona, Spain * Correspondence: [email protected] (S.J.F.); [email protected] (J.G.-C.) † Current address: Orchestra Scientific S.L. Av. Paisos Catalans, 16, 43007 Tarragona, Spain. Abstract: Energy production and consumption without the use of fossil fuels are amongst the biggest challenges currently facing humankind and the scientific community. Huge efforts have been invested in creating technologies that enable closed carbon or carbon neutral fuel cycles, limiting CO2 emissions into the atmosphere. Formic acid/formate (FA) has attracted intense interest as a liquid fuel over the last half century, giving rise to a plethora of studies on catalysts for its efficient electrocatalytic oxidation for usage in fuel cells. However, new catalysts and catalytic systems are often difficult to compare because of the variability in conditions and catalyst parameters examined. In this review, we Citation: Folkman, S.J.; discuss the extensive literature on FA electrooxidation using platinum, palladium and non-platinum González-Cobos, J.; Giancola, S.; group metal-based catalysts, the conditions typically employed in formate electrooxidation and the Sánchez-Molina, I.; Galán-Mascarós, main electrochemical parameters for the comparison of anodic electrocatalysts to be applied in a FA J.R. -

Microfabricated Solid Oxide Fuel Cells
Microfabricated Solid Oxide Fuel Cells THÈSE NO 4175 (2008) PRÉSENTÉE LE 26 SEPTEMBRE 2008 À LA FACULTE SCIENCES ET TECHNIQUES DE L'INGÉNIEUR LABORATOIRE DE CÉRAMIQUE PROGRAMME DOCTORAL EN SCIENCE ET GÉNIE DES MATÉRIAUX ÉCOLE POLYTECHNIQUE FÉDÉRALE DE LAUSANNE POUR L'OBTENTION DU GRADE DE DOCTEUR ÈS SCIENCES PAR Samuel REY-MERMET ingénieur en science des matériaux diplômé EPF de nationalité suisse et originaire de Val-d'Illiez (VS) acceptée sur proposition du jury: Dr N. Baluc, présidente du jury Prof. P. Muralt, directeur de thèse Prof. L. Gauckler, rapporteur Dr P. Hoffmann, rapporteur Dr H.-P. Loebl, rapporteur Suisse 2008 i Abstract Micro-fabricated solid oxide fuel cells (μSOFCs) are finding an increasing interest as potential power sources for portable devices such as MP3 players or laptops. The aim of this work was to fabricate a μSOFC demonstrator that works at 500°C and is fuelled by hydrogen. This thesis was divided into two parts. The first one was devoted to the development of an electrolyte and electrodes in form of sputtered thin films with electrical and mechanical properties suitable for the implementation in a real cell. YSZ and CGO electrolyte have been reactively sputtered from metallic targets. Both films are dense and have a columnar microstructure. The ionic conductivity of these films was of 0.5 S/m at 550°C for the CGO and of 5.5 x 10-3 S/m at 500°C for the yttria stabilized zirconia (YSZ) . Albeit the ceria doped gadolinia (CGO) was a better ionic conductor at low temperature, it was not possible to obtain an open circuit voltage (OCV) with a CGO electrolyte film.