S42003-021-02510-6.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Diversity of Understudied Archaeal and Bacterial Populations of Yellowstone National Park: from Genes to Genomes Daniel Colman
University of New Mexico UNM Digital Repository Biology ETDs Electronic Theses and Dissertations 7-1-2015 Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes Daniel Colman Follow this and additional works at: https://digitalrepository.unm.edu/biol_etds Recommended Citation Colman, Daniel. "Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes." (2015). https://digitalrepository.unm.edu/biol_etds/18 This Dissertation is brought to you for free and open access by the Electronic Theses and Dissertations at UNM Digital Repository. It has been accepted for inclusion in Biology ETDs by an authorized administrator of UNM Digital Repository. For more information, please contact [email protected]. Daniel Robert Colman Candidate Biology Department This dissertation is approved, and it is acceptable in quality and form for publication: Approved by the Dissertation Committee: Cristina Takacs-Vesbach , Chairperson Robert Sinsabaugh Laura Crossey Diana Northup i Diversity of understudied archaeal and bacterial populations from Yellowstone National Park: from genes to genomes by Daniel Robert Colman B.S. Biology, University of New Mexico, 2009 DISSERTATION Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biology The University of New Mexico Albuquerque, New Mexico July 2015 ii DEDICATION I would like to dedicate this dissertation to my late grandfather, Kenneth Leo Colman, associate professor of Animal Science in the Wool laboratory at Montana State University, who even very near the end of his earthly tenure, thought it pertinent to quiz my knowledge of oxidized nitrogen compounds. He was a man of great curiosity about the natural world, and to whom I owe an acknowledgement for his legacy of intellectual (and actual) wanderlust. -

Expanding the Chlamydiae Tree
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 2040 Expanding the Chlamydiae tree Insights into genome diversity and evolution JENNAH E. DHARAMSHI ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6214 ISBN 978-91-513-1203-3 UPPSALA urn:nbn:se:uu:diva-439996 2021 Dissertation presented at Uppsala University to be publicly examined in A1:111a, Biomedical Centre (BMC), Husargatan 3, Uppsala, Tuesday, 8 June 2021 at 13:15 for the degree of Doctor of Philosophy. The examination will be conducted in English. Faculty examiner: Prof. Dr. Alexander Probst (Faculty of Chemistry, University of Duisburg-Essen). Abstract Dharamshi, J. E. 2021. Expanding the Chlamydiae tree. Insights into genome diversity and evolution. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 2040. 87 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-513-1203-3. Chlamydiae is a phylum of obligate intracellular bacteria. They have a conserved lifecycle and infect eukaryotic hosts, ranging from animals to amoeba. Chlamydiae includes pathogens, and is well-studied from a medical perspective. However, the vast majority of chlamydiae diversity exists in environmental samples as part of the uncultivated microbial majority. Exploration of microbial diversity in anoxic deep marine sediments revealed diverse chlamydiae with high relative abundances. Using genome-resolved metagenomics various marine sediment chlamydiae genomes were obtained, which significantly expanded genomic sampling of Chlamydiae diversity. These genomes formed several new clades in phylogenomic analyses, and included Chlamydiaceae relatives. Despite endosymbiosis-associated genomic features, hosts were not identified, suggesting chlamydiae with alternate lifestyles. Genomic investigation of Anoxychlamydiales, newly described here, uncovered genes for hydrogen metabolism and anaerobiosis, suggesting they engage in syntrophic interactions. -

Compendium of Measures to Control Chlamydia Psittaci Infection Among
Compendium of Measures to Control Chlamydia psittaci Infection Among Humans (Psittacosis) and Pet Birds (Avian Chlamydiosis), 2017 Author(s): Gary Balsamo, DVM, MPH&TMCo-chair Angela M. Maxted, DVM, MS, PhD, Dipl ACVPM Joanne W. Midla, VMD, MPH, Dipl ACVPM Julia M. Murphy, DVM, MS, Dipl ACVPMCo-chair Ron Wohrle, DVM Thomas M. Edling, DVM, MSpVM, MPH (Pet Industry Joint Advisory Council) Pilar H. Fish, DVM (American Association of Zoo Veterinarians) Keven Flammer, DVM, Dipl ABVP (Avian) (Association of Avian Veterinarians) Denise Hyde, PharmD, RP Preeta K. Kutty, MD, MPH Miwako Kobayashi, MD, MPH Bettina Helm, DVM, MPH Brit Oiulfstad, DVM, MPH (Council of State and Territorial Epidemiologists) Branson W. Ritchie, DVM, MS, PhD, Dipl ABVP, Dipl ECZM (Avian) Mary Grace Stobierski, DVM, MPH, Dipl ACVPM (American Veterinary Medical Association Council on Public Health and Regulatory Veterinary Medicine) Karen Ehnert, and DVM, MPVM, Dipl ACVPM (American Veterinary Medical Association Council on Public Health and Regulatory Veterinary Medicine) Thomas N. Tully JrDVM, MS, Dipl ABVP (Avian), Dipl ECZM (Avian) (Association of Avian Veterinarians) Source: Journal of Avian Medicine and Surgery, 31(3):262-282. Published By: Association of Avian Veterinarians https://doi.org/10.1647/217-265 URL: http://www.bioone.org/doi/full/10.1647/217-265 BioOne (www.bioone.org) is a nonprofit, online aggregation of core research in the biological, ecological, and environmental sciences. BioOne provides a sustainable online platform for over 170 journals and books published by nonprofit societies, associations, museums, institutions, and presses. Your use of this PDF, the BioOne Web site, and all posted and associated content indicates your acceptance of BioOne’s Terms of Use, available at www.bioone.org/page/terms_of_use. -

Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline
Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline • Phlogenetics of Archaea • Phlogenetics of archaeal lipids • Papers Phyla • Two? main phyla – Euryarchaeota • Methanogens • Extreme halophiles • Extreme thermophiles • Sulfate-reducing – Crenarchaeota • Extreme thermophiles – Korarchaeota? • Hyperthermophiles • indicated only by environmental DNA sequences – Nanoarchaeum? • N. equitans a fast evolving euryarchaeal lineage, not novel, early diverging archaeal phylum – Ancient archael group? • In deepest brances of Crenarchaea? Euryarchaea? Archaeal Lipids • Methanogens – Di- and tetra-ethers of glycerol and isoprenoid alcohols – Core mostly archaeol or caldarchaeol – Core sometimes sn-2- or Images removed due to sn-3-hydroxyarchaeol or copyright considerations. macrocyclic archaeol –PMI • Halophiles – Similar to methanogens – Exclusively synthesize bacterioruberin • Marine Crenarchaea Depositional Archaeal Lipids Biological Origin Environment Crocetane methanotrophs? methane seeps? methanogens, PMI (2,6,10,15,19-pentamethylicosane) methanotrophs hypersaline, anoxic Squalane hypersaline? C31-C40 head-to-head isoprenoids Smit & Mushegian • “Lost” enzymes of MVA pathway must exist – Phosphomevalonate kinase (PMK) – Diphosphomevalonate decarboxylase – Isopentenyl diphosphate isomerase (IPPI) Kaneda et al. 2001 Rohdich et al. 2001 Boucher et al. • Isoprenoid biosynthesis of archaea evolved through a combination of processes – Co-option of ancestral enzymes – Modification of enzymatic specificity – Orthologous and non-orthologous gene -

Chlamydophila Felis Prevalence Chlamydophila Felis Prevalence
ORIGINAL SCIENTIFIC ARTICLE / IZVORNI ZNANSTVENI ČLANAK A preliminary study of Chlamydophila felis prevalence among domestic cats in the City of Zagreb and Zagreb County in Croatia Gordana Gregurić Gračner*, Ksenija Vlahović, Alenka Dovč, Brigita Slavec, Ljiljana Bedrica, S. Žužul and D. Gračner Introduction Feline chlamydiosis is a disease in do- Rampazzo et al. (2003) investigated mestic cats caused by Chlamydophila felis the prevalence of Cp. felis and feline (Cp. felis), which is primarily a pathogen herpesvirus in cats with conjunctivitis by of the conjunctiva and nasal mucosa using a conventional polymerase chain rather than a pulmonary pathogen. It is reaction (PCR), and discovered that 14 capable of causing acute to chronic con- out of 70 (20%) cats with conjunctivitis junctivitis, with blepharospasm, chem- were positive only on Cp. felis and mixed osis and congestion, a serous to mucop- infections with herpesvirus were present urulent ocular discharge, and rhinitis in 5 of 70 (7%) cats. (Hoover et al., 1978; Sykes, 2005). C. Helps et al. (2005) took oropharyngeal 1 psittaci infection in kittens produces fe- and conjunctival swabs from 1101 cats ver, lethargy, lameness, and reduction and by using a PCR determined Cp. felis in weight gain (Terwee at al., 1998). Ac- in 10% of the 558 swab samples of cats cording to the literature, chlamydiosis with URDT and in 3% of the 558 swab in cats can be treated successfully by samples of cats without URDT. administering potentiated amoxicillin Low et al. (2007) investigated 55 cats for 30 days, which can result in a com- with conjunctivitis, 39 healthy cats and plete clinical recovery with no evidence 32 cats with a history of conjunctivitis of a recurrence for six months (Sturgess that been resolved for at least 3 months. -

Product Description EN Bactoreal® Kit Chlamydiaceae
Product Description BactoReal® Kit Chlamydiaceae For research only, not for diagnostic use BactoReal® Kit Chlamydiaceae Order no. Reactions Pathogen Internal positive control DVEB03113 100 FAM channel Cy5 channel DVEB03153 50 FAM channel Cy5 channel DVEB03111 100 FAM channel VIC/HEX channel DVEB03151 50 FAM channel VIC/HEX channel Kit contents: Detection assay for Chlamydia and Chlamydophila species Detection assay for internal positive control (control of amplification) DNA reaction mix (contains uracil-N glycosylase, UNG) Positive control for Chlamydiaceae Water Background: The Chlamydiaceae family currently includes two genera and one candidate genus: genus Chlamydia (including the species Chlamydia muridarum, Chlamydia suis, Chlamydia trachomatis), genus Chlamydophila (including the species Chlamydophila abortus, Chlamydophila caviae Chlamydophila felis, Chlamydia pecorum, Chlamydophila pneumoniae, Chlamydophila psittaci), and candidatus Clavochlamydia. All Chlamydiaceae are obligate intracellular Gram-negative bacteria that cause diseases in humans and animals worldwide. Description: BactoReal® Kit Chlamydiaceae is based on the amplification and detection of the 23S rRNA gene of C. muridarum, C. suis, C. trachomatis, C. abortus, C. caviae, C. felis, C. pecorum, C. pneumoniae and C. psittaci using real-time PCR. It allows the rapid and sensitive detection of the 23S rRNA gene of Chlamydiaceae from DNA samples purified from different sample material (e.g. with the QIAamp DNA Mini Kit). For subtyping please contact ingenetix. PCR-platforms: BactoReal® Kit Chlamydiaceae is developed and validated for the ABI PRISM® 7500 instrument (Life Technologies), LightCycler® 480 (Roche) and Mx3005P® QPCR System (Agilent), but is also suitable for other real-time PCR instruments. Sensitivity and specificity: BactoReal® Kit Chlamydiaceae detects at least 10 copies/reaction. -
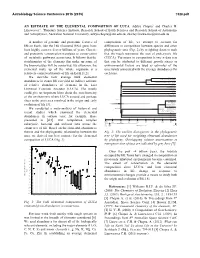
An Estimate of the Elemental Composition of Luca
Astrobiology Science Conference 2015 (2015) 7328.pdf AN ESTIMATE OF THE ELEMENTAL COMPOSITION OF LUCA. Aditya Chopra1 and Charles H. Lineweaver1, 1Planetary Science Institute, Research School of Earth Sciences and Research School of Astronomy and Astrophysics, Australian National University, [email protected], [email protected] A number of genomic and proteomic features of composition of life, we attempt to account for life on Earth, like the 16S ribosomal RNA gene, have differences in composition between species and other been highly conserved over billions of years. Genetic phylogenetic taxa (Fig. 2) by weighting datasets such and proteomic conservation translates to conservation that the result represents the root of prokaryotic life of metabolic pathways across taxa. It follows that the (LUCA). Variations in composition between data sets stoichiometry of the elements that make up some of that can be attributed to different growth stages or the biomolecules will be conserved. By extension, the environmental factors are used as estimates of the elemental make up of the whole organism is a uncertainty associated with the average abundances for relatively conserved feature of life on Earth [1,2]. each taxa. We describe how average bulk elemental Euryarchaeota 1a Methanococci, Methanobacteria, Methanopyri 7 Euryarchaeota 1b abundances in extant life can yield an indirect estimate 6 Thermoplasmata, Methanomicrobia, Halobacteria, Archaeoglobi Euryarchaeota 2 4 Thermococci of relative abundances of elements in the Last Crenarchaeota Sulfolobus, Thermoproteus ? Thaumarchaeota Universal Common Ancestor (LUCA). The results Cenarchaeum ? Korarchaeota ARMAN could give us important hints about the stoichiometry ? 2 Archaeal Richmond Mine Acidophilic Nanoorganisms Nanoarchaeota of the environment where LUCA existed and perhaps Archaea Terrabacteria clues to the processes involved in the origin and early Actinobacteria, Deinococcus-Thermus, 1 Cyanobacteria, Life Chloroflexi, 9 evolution of life [3]. -

Pan-Genome Analysis and Ancestral State Reconstruction Of
www.nature.com/scientificreports OPEN Pan‑genome analysis and ancestral state reconstruction of class halobacteria: probability of a new super‑order Sonam Gaba1,2, Abha Kumari2, Marnix Medema 3 & Rajeev Kaushik1* Halobacteria, a class of Euryarchaeota are extremely halophilic archaea that can adapt to a wide range of salt concentration generally from 10% NaCl to saturated salt concentration of 32% NaCl. It consists of the orders: Halobacteriales, Haloferaciales and Natriabales. Pan‑genome analysis of class Halobacteria was done to explore the core (300) and variable components (Softcore: 998, Cloud:36531, Shell:11784). The core component revealed genes of replication, transcription, translation and repair, whereas the variable component had a major portion of environmental information processing. The pan‑gene matrix was mapped onto the core‑gene tree to fnd the ancestral (44.8%) and derived genes (55.1%) of the Last Common Ancestor of Halobacteria. A High percentage of derived genes along with presence of transformation and conjugation genes indicate the occurrence of horizontal gene transfer during the evolution of Halobacteria. A Core and pan‑gene tree were also constructed to infer a phylogeny which implicated on the new super‑order comprising of Natrialbales and Halobacteriales. Halobacteria1,2 is a class of phylum Euryarchaeota3 consisting of extremely halophilic archaea found till date and contains three orders namely Halobacteriales4,5 Haloferacales5 and Natrialbales5. Tese microorganisms are able to dwell at wide range of salt concentration generally from 10% NaCl to saturated salt concentration of 32% NaCl6. Halobacteria, as the name suggests were once considered a part of a domain "Bacteria" but with the discovery of the third domain "Archaea" by Carl Woese et al.7, it became part of Archaea. -

Insight in the Biology of Chlamydia-Related Bacteria
+ MODEL Microbes and Infection xx (2017) 1e9 www.elsevier.com/locate/micinf Insight in the biology of Chlamydia-related bacteria Firuza Bayramova, Nicolas Jacquier, Gilbert Greub* Centre for Research on Intracellular Bacteria, Institute of Microbiology, University Hospital Centre and University of Lausanne, Bugnon 48, 1011 Lausanne, Switzerland Received 26 September 2017; accepted 21 November 2017 Available online ▪▪▪ Abstract The Chlamydiales order is composed of obligate intracellular bacteria and includes the Chlamydiaceae family and several family-level lineages called Chlamydia-related bacteria. In this review we will highlight the conserved and distinct biological features between these two groups. We will show how a better characterization of Chlamydia-related bacteria may increase our understanding on the Chlamydiales order evolution, and may help identifying new therapeutic targets to treat chlamydial infections. © 2017 The Author(s). Published by Elsevier Masson SAS on behalf of Institut Pasteur. This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/). Keywords: Chlamydiae; Chlamydia-related bacteria; Phylogeny; Chlamydial division 1. Introduction Data indicate that most of the members of this order might be pathogenic for humans and/or animals [4,18e23, The Chlamydiales order, composed of Gram-negative 11,12,24,16].TheChlamydiaceae are currently the best obligate intracellular bacteria, shares a unique biphasic described family of the Chlamydiales order [25]. The Chla- developmental cycle. The order includes important pathogens mydiaceae family is composed of 11 species including three of humans and animals. The order Chlamydiales includes the major human and several animal pathogens. Chlamydiaceae family and several family-level lineages Chlamydia trachomatis is the most common sexually collectively called Chlamydia-related bacteria. -

CHLAMYDIOSIS (Psittacosis, Ornithosis)
EAZWV Transmissible Disease Fact Sheet Sheet No. 77 CHLAMYDIOSIS (Psittacosis, ornithosis) ANIMAL TRANS- CLINICAL FATAL TREATMENT PREVENTION GROUP MISSION SIGNS DISEASE ? & CONTROL AFFECTED Birds Aerogenous by Very species Especially the Antibiotics, Depending on Amphibians secretions and dependent: Chlamydophila especially strain. Reptiles excretions, Anorexia psittaci is tetracycline Mammals Dust of Apathy ZOONOSIS. and In houses People feathers and Dispnoe Other strains doxycycline. Maximum of faeces, Diarrhoea relative host For hygiene in Oral, Cachexy specific. substitution keeping and Direct Conjunctivitis electrolytes at feeding. horizontal, Rhinorrhea Yes: persisting Vertical, Nervous especially in diarrhoea. in zoos By parasites symptoms young animals avoid stress, (but not on the Reduced and animals, quarantine, surface) hatching rates which are blood screening, Increased new- damaged in any serology, born mortality kind. However, take swabs many animals (throat, cloaca, are carrier conjunctiva), without clinical IFT, PCR. symptoms. Fact sheet compiled by Last update Werner Tschirch, Veterinary Department, March 2002 Hoyerswerda, Germany Fact sheet reviewed by E. F. Kaleta, Institution for Poultry Diseases, Justus-Liebig-University Gießen, Germany G. M. Dorrestein, Dept. Pathology, Utrecht University, The Netherlands Susceptible animal groups In case of Chlamydophila psittaci: birds of every age; up to now proved in 376 species of birds of 29 birds orders, including 133 species of parrots; probably all of the about 9000 species of birds are susceptible for the infection; for the outbreak of the disease, additional factors are necessary; very often latent infection in captive as well as free-living birds. Other susceptible groups are amphibians, reptiles, many domestic and wild mammals as well as humans. The other Chlamydia sp. -

Psittacosis/ Avian Chlamydiosis
Psittacosis/ Importance Avian chlamydiosis, which is also called psittacosis in some hosts, is a bacterial Avian disease of birds caused by members of the genus Chlamydia. Chlamydia psittaci has been the primary organism identified in clinical cases, to date, but at least two Chlamydiosis additional species, C. avium and C. gallinacea, have now been recognized. C. psittaci is known to infect more than 400 avian species. Important hosts among domesticated Ornithosis, birds include psittacines, poultry and pigeons, but outbreaks have also been Parrot Fever documented in many other species, such as ratites, peacocks and game birds. Some individual birds carry C. psittaci asymptomatically. Others become mildly to severely ill, either immediately or after they have been stressed. Significant economic losses Last Updated: April 2017 are possible in commercial turkey flocks even when mortality is not high. Outbreaks have been reported occasionally in wild birds, and some of these outbreaks have been linked to zoonotic transmission. C. psittaci can affect mammals, including humans, that have been exposed to birds or contaminated environments. Some infections in people are subclinical; others result in mild to severe illnesses, which can be life-threatening. Clinical cases in pregnant women may be especially severe, and can result in the death of the fetus. Recent studies suggest that infections with C. psittaci may be underdiagnosed in some populations, such as poultry workers. There are also reports suggesting that it may occasionally cause reproductive losses, ocular disease or respiratory illnesses in ruminants, horses and pets. C. avium and C. gallinacea are still poorly understood. C. avium has been found in asymptomatic pigeons, which seem to be its major host, and in sick pigeons and psittacines. -

Seroprevalence and Risk Factors Associated with Chlamydia Abortus Infection in Sheep and Goats in Eastern Saudi Arabia
pathogens Article Seroprevalence and Risk Factors Associated with Chlamydia abortus Infection in Sheep and Goats in Eastern Saudi Arabia Mahmoud Fayez 1,2,† , Ahmed Elmoslemany 3,† , Mohammed Alorabi 4, Mohamed Alkafafy 4 , Ibrahim Qasim 5, Theeb Al-Marri 1 and Ibrahim Elsohaby 6,7,*,† 1 Al-Ahsa Veterinary Diagnostic Lab, Ministry of Environment, Water and Agriculture, Al-Ahsa 31982, Saudi Arabia; [email protected] (M.F.); [email protected] (T.A.-M.) 2 Department of Bacteriology, Veterinary Serum and Vaccine Research Institute, Ministry of Agriculture, Cairo 131, Egypt 3 Hygiene and Preventive Medicine Department, Faculty of Veterinary Medicine, Kafrelsheikh University, Kafr El-Sheikh 33516, Egypt; [email protected] 4 Department of Biotechnology, College of Science, Taif University, P.O. Box 11099, Taif 21944, Saudi Arabia; [email protected] (M.A.); [email protected] (M.A.) 5 Department of Animal Resources, Ministry of Environment, Water and Agriculture, Riyadh 12629, Saudi Arabia; [email protected] 6 Department of Animal Medicine, Faculty of Veterinary Medicine, Zagazig University, Zagazig City 44511, Egypt 7 Department of Health Management, Atlantic Veterinary College, University of Prince Edward Island, Charlottetown, PE C1A 4P3, Canada * Correspondence: [email protected]; Tel.: +1-902-566-6063 † These authors contributed equally to this work. Citation: Fayez, M.; Elmoslemany, Abstract: Chlamydia abortus (C. abortus) is intracellular, Gram-negative bacterium that cause enzootic A.; Alorabi, M.; Alkafafy, M.; Qasim, abortion in sheep and goats. Information on C. abortus seroprevalence and flock management risk I.; Al-Marri, T.; Elsohaby, I. factors associated with C. abortus seropositivity in sheep and goats in Saudi Arabia are scarce.