Increasing Complexity of Ras Signaling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

REVIEW Signal Transduction, Cell Cycle Regulatory, and Anti
Leukemia (1999) 13, 1109–1166 1999 Stockton Press All rights reserved 0887-6924/99 $12.00 http://www.stockton-press.co.uk/leu REVIEW Signal transduction, cell cycle regulatory, and anti-apoptotic pathways regulated by IL-3 in hematopoietic cells: possible sites for intervention with anti-neoplastic drugs WL Blalock1, C Weinstein-Oppenheimer1,2, F Chang1, PE Hoyle1, X-Y Wang3, PA Algate4, RA Franklin1,5, SM Oberhaus1,5, LS Steelman1 and JA McCubrey1,5 1Department of Microbiology and Immunology, 5Leo Jenkins Cancer Center, East Carolina University School of Medicine Greenville, NC, USA; 2Escuela de Quı´mica y Farmacia, Facultad de Medicina, Universidad de Valparaiso, Valparaiso, Chile; 3Department of Laboratory Medicine and Pathology, Mayo Clinic and Foundation, Rochester, MN, USA; and 4Division of Basic Sciences, Fred Hutchinson Cancer Research Center, Seattle, WA, USA Over the past decade, there has been an exponential increase growth factor), Flt-L (the ligand for the flt2/3 receptor), erythro- in our knowledge of how cytokines regulate signal transduc- poietin (EPO), and others affect the growth and differentiation tion, cell cycle progression, differentiation and apoptosis. Research has focused on different biochemical and genetic of these early hematopoietic precursor cells into cells of the 1–4 aspects of these processes. Initially, cytokines were identified myeloid, lymphoid and erythroid lineages (Table 1). This by clonogenic assays and purified by biochemical techniques. review will concentrate on IL-3 since much of the knowledge This soon led to the molecular cloning of the genes encoding of how cytokines affect cell growth, signal transduction, and the cytokines and their cognate receptors. -

Autophagy: from Basic Science to Clinical Application
nature publishing group REVIEW See COMMENTARY page XX Autophagy: from basic science to clinical application J Va n L i m b e r g e n 1 , 2 , 3 , C S t e v e n s 4 , E R N i m m o 1 , D C W i l s o n 2 , 3 a n d J S a t s a n g i 1 Autophagy is a cellular pathway involved in protein and organelle degradation, which is likely to represent an innate adaptation to starvation. In times of nutrient deficiency, the cell can self-digest and recycle some nonessential components through nonselective autophagy, thus sustaining minimal growth requirements until a food source becomes available. Over recent years, autophagy has been implicated in an increasing number of clinical scenarios, notably infectious diseases, cancer, neurodegenerative diseases, and autoimmunity. The recent identification of the importance of autophagy genes in the genetic susceptibility to Crohn ’ s disease suggests that a selective autophagic response may play a crucial role in the pathogenesis of common complex immune-mediated diseases. In this review, we discuss the autophagic mechanisms, their molecular regulation, and summarize their clinical relevance. This progress has led to great interest in the therapeutic potential of manipulation of both selective and nonselective autophagy in established disease. INTRODUCTION The ability to adapt to environmental change is essential for sur- Autophagy encompasses several distinct processes involving vival. This is true for the organism as a whole and for individual the delivery of portions of the cytoplasm to the lysosome for cells alike. -

Characterization of Gf a Drosophila Trimeric G Protein Alpha Subunit
Characterization of Gf a Drosophila trimeric G protein alpha subunit Naureen Quibria Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2012 © 2012 Naureen Quibria All rights reserved Abstract Characterization of Gf a Drosophila trimeric G-protein alpha subunit Naureen Quibria In the morphogenesis of tissue development, how coordination of patterning and growth achieve the correct organ size and shape is a principal question in biology. Efficient orchestrating mechanisms are required to achieve this and cells have developed sophisticated systems for reception and interpretation of the multitude of extracellular stimuli to which they are exposed. Plasma membrane receptors play a key role in the transmission of such signals. G-protein coupled receptors (GPCRs) are the largest class of cell surface receptors that respond to an enormous diversity of extracellular stimuli, and are critical mediators of cellular signal transduction in eukaryotic organisms. Signaling through GPCRs has been well characterized in many biological contexts. While they are a major class of signal transducers, there are not many defined instances where GPCRs have been implicated in the process of development to date. The Drosophila wing provides an ideal model system to elucidate and address the role of GPCRs in development, as its growth is regulated by a small number of conserved signaling pathways. In my thesis work, I address the role of a trimeric G alpha protein in Drosophila, Gαf, and what part it may play in development. In particular, I explore the role of Gαf as an alpha subunit of a trimeric complex, to determine what heptahelical receptors might act as its cognate receptor. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Phosphorylation of Chicken Protein Tyrosine Phosphatase 1 by Casein Kinase II in Vitro
EXPERIMENTAL and MOLECULAR MEDICINE, Vol. 29, No 4, 229-233, December 1997 Phosphorylation of chicken protein tyrosine phosphatase 1 by casein kinase II in vitro Eun Joo Jung,1 Kee Ryeon Kang1 and Introduction Yoon-Se Kang1,2 The phosphorylation of protein tyrosine residues is an early event in signal transduction initiated by binding of 1 Department of Biochemistry and Gyeongsang Institute of Cancer growth factors and hormones to their cognate receptors Research, College of Medicine, Gyeongsang National University, and it leads to regulation of cellular activities which include Chinju 660-280, Korea proliferation, differentiation, and also malignant transfor- 2 Corresponding author mation of cells (Hunter, 1989; Ullirich and Schlessinger, Accepted 17 November 1997 1990; Cantley et al., 1991). Under normal conditions, the level of tyrosine phosphorylation within a cell is determined by a balance between the actions of protein tyrosine Abbreviations: CPTP, chicken protein tyrosine phosphatase; HPTP1B, human placenta kinases (PTKs) and protein tyrosine phosphatases (PTPs) protein tyrosine phosphatase 1B; CKII, casein kinase II; MAP kinase, mitogen-activated (Hunter, 1989; Fischer et al., 1991; Trowbridge, 1991). protein kinase; GST, glutathione S-transferase; pNPP, p-nitrophenyl phosphate; EGF, PTPs do not simply reverse the action of tyrosine kinases, epidermal growth factor but rather, PTP itself may play a central role in cellular regulation. PTPs are generally classified as transmem- brane (receptor-type) and nontransmembrane (nonrecep- tor-type) enzymes based on the presence or absence of extracellular and transmembrane portions of their predicted sequence (Fischer et al., 1991). Because the activity of Abstract tyrosine kinase can be controlled by phosphorylation, it has been postulated that PTP activity may be regulated The phosphorylation and dephosphorylation of by phosphorylation as well. -

Mechanism of Insulin Receptor Kinase Inhibition in Non-Insulin- Dependent Diabetes Mellitus Patients
Mechanism of insulin receptor kinase inhibition in non-insulin- dependent diabetes mellitus patients. Phosphorylation of serine 1327 or threonine 1348 is unaltered. M Kellerer, … , K Siddle, H U Häring J Clin Invest. 1995;96(1):6-11. https://doi.org/10.1172/JCI118073. Research Article The tyrosine kinase activity of insulin receptor isolated from the skeletal muscle of NIDDM patients has previously been found to be decreased compared with the activity of receptor from nondiabetic subjects but the mechanism underlying this defect is unknown. Phosphorylation of receptor serine/threonine residues has been proposed to exert an inhibitory influence on receptor tyrosine kinase activity and Ser 1327 and Thr 1348 have been identified as specific sites of phosphorylation in the insulin receptor COOH terminal domain. To address the potential negative regulatory role of phosphorylation of these residues in vivo, we assessed the extent of phosphorylation of each site in insulin receptor isolated from the skeletal muscle of 12 NIDDM patients and 13 nondiabetic, control subjects. Phosphorylation of Ser 1327 and Thr 1348 was determined using antibodies that specifically recognize insulin receptor phosphorylated at these sites. In addition, a phosphotyrosine-specific antibody was used to monitor receptor tyrosine phosphorylation. The extent of insulin-induced tyrosine autophosphorylation was decreased in receptor isolated from diabetic versus nondiabetic muscle, thus confirming earlier reports. In contrast, there was no significant difference in the extent of phosphorylation of either Ser 1327 or Thr 1348 in receptor isolated from diabetic or nondiabetic muscle as assessed by immunoprecipitation (Ser 1327: 5.6 +/- 1.6% diabetics vs. 4.7 +/- 2.0% control; Thr 1348: 3.8 +/- 1.0% diabetics vs. -

Hras Intracellular Trafficking and Signal Transduction Jodi Ho-Jung Mckay Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 2007 HRas intracellular trafficking and signal transduction Jodi Ho-Jung McKay Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Biological Phenomena, Cell Phenomena, and Immunity Commons, Cancer Biology Commons, Cell Biology Commons, Genetics and Genomics Commons, and the Medical Cell Biology Commons Recommended Citation McKay, Jodi Ho-Jung, "HRas intracellular trafficking and signal transduction" (2007). Retrospective Theses and Dissertations. 13946. https://lib.dr.iastate.edu/rtd/13946 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. HRas intracellular trafficking and signal transduction by Jodi Ho-Jung McKay A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Genetics Program of Study Committee: Janice E. Buss, Co-major Professor Linda Ambrosio, Co-major Professor Diane Bassham Drena Dobbs Ted Huiatt Iowa State University Ames, Iowa 2007 Copyright © Jodi Ho-Jung McKay, 2007. All rights reserved. UMI Number: 3274881 Copyright 2007 by McKay, Jodi Ho-Jung All rights reserved. UMI Microform 3274881 Copyright 2008 by ProQuest Information and Learning Company. All rights reserved. This microform edition is protected against unauthorized copying under Title 17, United States Code. ProQuest Information and Learning Company 300 North Zeeb Road P.O. -

G-Protein ␥-Complex Is Crucial for Efficient Signal Amplification in Vision
The Journal of Neuroscience, June 1, 2011 • 31(22):8067–8077 • 8067 Cellular/Molecular G-Protein ␥-Complex Is Crucial for Efficient Signal Amplification in Vision Alexander V. Kolesnikov,1 Loryn Rikimaru,2 Anne K. Hennig,1 Peter D. Lukasiewicz,1 Steven J. Fliesler,4,5,6,7 Victor I. Govardovskii,8 Vladimir J. Kefalov,1 and Oleg G. Kisselev2,3 1Department of Ophthalmology and Visual Sciences, Washington University School of Medicine, St. Louis, Missouri 63110, Departments of 2Ophthalmology and 3Biochemistry and Molecular Biology, Saint Louis University School of Medicine, Saint Louis, Missouri 63104, 4Research Service, Veterans Administration Western New York Healthcare System, and Departments of 5Ophthalmology (Ross Eye Institute) and 6Biochemistry, University at Buffalo/The State University of New York (SUNY), and 7SUNY Eye Institute, Buffalo, New York 14215, and 8Sechenov Institute for Evolutionary Physiology and Biochemistry, Russian Academy of Sciences, Saint Petersburg 194223, Russia A fundamental question of cell signaling biology is how faint external signals produce robust physiological responses. One universal mechanism relies on signal amplification via intracellular cascades mediated by heterotrimeric G-proteins. This high amplification system allows retinal rod photoreceptors to detect single photons of light. Although much is now known about the role of the ␣-subunit of the rod-specific G-protein transducin in phototransduction, the physiological function of the auxiliary ␥-complex in this process remains a mystery. Here, we show that elimination of the transducin ␥-subunit drastically reduces signal amplification in intact mouse rods. The consequence is a striking decline in rod visual sensitivity and severe impairment of nocturnal vision. Our findings demonstrate that transducin ␥-complex controls signal amplification of the rod phototransduction cascade and is critical for the ability of rod photoreceptors to function in low light conditions. -

Brief Definitive Report ZAP- 70 Gene
Brief Definitive Report Reconstitution of T Cell Receptor Signaling in ZAP-70-deficient Cells by Retroviral Transduction of the ZAP- 70 Gene By Naomi Taylor,* Kevin B. Bacon,¢ Susan Smith,* Thomas Jahn,* Theresa A. Kadlecekfl Lisa Uribe,* Donald B. Kohn,* Erwin W. Gelfand,IIArthur Weiss,~ and Kenneth Weinberg* From the *Division of Research Immunology and Bone Marrow Transplantation, Children's Hospital Los Angeles, Los Angeles, California 90027; :~DNAX Research Institute, Palo Alto, California 94304; gHoward Hughes Medical Institute, Department of Medicine and of Microbiology and Immunology, University of California, San Francisco, California 94143; and IIDivision of Basic Sciences and Molecular Signal Transduction Program, Department of Pediatrics, National Jewish Centerfor Immunology and Respiratory Diseases, Denver, Colorado 80206 Summal-y A variant of severe combined lmmunodeficiency syndrome (SCID) with a selective inability to produce CD8 single positive T cells and a signal transduction defect in peripheral CD4 + cells has recently been shown to be the result of mutations in the ZAP-70 gene. T cell receptor (TCR) signaling requires the association of the ZAP-70 protein tyrosine kinase with the TCR complex. Human T cell leukemia virus type I-transformed CD4 + T cell lines w.ere established from ZAP-70-deficient patients and normal controls. ZAP-70 was expressed and appropriately phosphorylated in normal T cell lines after TCR engagement, but was not detected in T cell lines from ZAP-70-deficient patients. To determine whether signaling could be reconstituted, wild-type ZAP-70 was introduced into deficient cells with a ZAP-70 retroviral vector. High titer producer clones expressing ZAP-70 were generated in the Gibbon ape leukemia virus packaging line PG13. -

Phosphorylation of Protein 4.1 on Tyrosine-418 Modulates Its Function
Proc. Nati. Acad. Sci. USA Vol. 88, pp. 5222-5226, June 1991 Biochemistry Phosphorylation of protein 4.1 on tyrosine-418 modulates its function in vitro (epidermal growth factor receptor/spectrin/actin assembly) GOSUKONDA SUBRAHMANYAM*, PAUL J. BERTICStt, AND RICHARD A. ANDERSON**§ Department of *Pharmacology and tPhysiological Chemistry, and the tCell and Molecular Biology Program, University of Wisconsin Medical School, 1300 University Avenue, Madison, WI 53706 Communicated by Vincent T. Marchesi, March IS, 1991 ABSTRACT Protein 4.1 was initially characterized as a actin/protein 4.1 complex (12). However, phosphorylation of protein that regulates cytoskeletal assembly in erythrocytes. protein 4.1 by protein kinase C also reduces protein 4.1 However, recent studies have shown that protein 4.1 is ubiq- binding to band 3 but not to glycophorin, while phosphory- uitous in mammalian cells. Here, we show that protein 4.1 is lation by cAMP-dependent kinase does not affect membrane phosphorylated on tyrosine by the epidermal growth factor interactions (12). These results suggest that protein 4.1 is receptor (EGFR) tyrosine kinase. The phosphorylation site has phosphorylated at multiple sites by different protein kinases, been localized to the 8-kDa domain, which has one tyrosine, and each phosphorylation event selectively modulates pro- tyrosine-418. The 8-kDa region is required for the assembly of tein 4.1 function. The phosphorylation sites by cAMP- the spectrin/actin complex, and phosphorylation by EGFR dependent protein kinase and protein kinase C are in the reduced the ability ofprotein 4.1 to promote the assembly ofthe 8-kDa and 16-kDa domains (10). -
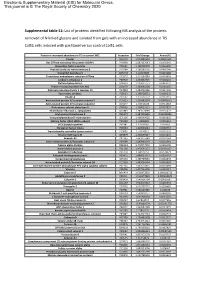
Supplemental Table S1
Electronic Supplementary Material (ESI) for Molecular Omics. This journal is © The Royal Society of Chemistry 2020 Supplemental table S1: List of proteins identified following MS analysis of the proteins removed of N-linked glycans and isolated from gels with an increased abundance in TIS Cal51 cells induced with paclitaxel versus control Cal51 cells. Protein in increased abundance in TIS vs control WCL Accession Fold Change Anova (P) Plectin Q15149 1.073855593 0.00691631 Ras GTPase-activating-like protein IQGAP1 P46940 1.087337643 0.0176342 Elongation factor1-gamma P26641 1.138709703 0.0116496 Peptidyl-prolyl cis-transisomerase B P23284 1.188383105 0.0436246 Dipeptidyl peptidase 3 Q9NY33 1.20163605 0.0215448 Transitional endoplasmic reticulum ATPase P55072 1.214194884 0.0449691 Carbonic anhydrase 2 P00918 1.232852325 0.0158141 Clathrin heavy chain 1 Q00610 1.239621773 0.0463237 Protein transport protein Sec 31A O94979 1.263565104 0.0284155 Aldo-ketoreductase family 1 member C1 Q04828 1.282092186 0.0324406 Spermidine synthase P19623 1.298728621 0.0196232 Plastin-3 P13797 1.310756772 0.0161319 Actin-related protein 2/3 complex subunit 5 O15511 1.333483524 0.00476923 Actin-related protein 2/3 complex subunit 2 O15144 1.35416168 0.0411018 Proteasome subunit alpha type-5 P28066 1.358015551 0.0337657 Thioredoxin reductase 1, cytoplasmic Q16881 1.383670089 0.0235472 Acyl-protein thioesterase 2 O95372 1.387415589 0.00233899 Isoaspartylpeptidase/L-asparaginase Q7L266 1.408149002 0.0319602 Splicing factor U2AF 65kDa subunit P26368 1.41489991 0.0256619 -

Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/Mtor Cascade Inhibitors: How Mutations Can Result in Therapy Resistance and How to Overcome Resistance
www.impactjournals.com/oncotarget/ Oncotarget, October, Vol.3, No 10 Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Cascade Inhibitors: How Mutations Can Result in Therapy Resistance and How to Overcome Resistance James A. McCubrey1, Linda S. Steelman1, William H. Chappell1, Stephen L. Abrams1, Richard A. Franklin1, Giuseppe Montalto2, Melchiorre Cervello3, Massimo Libra4, Saverio Candido4, Grazia Malaponte4, Maria C. Mazzarino4, Paolo Fagone4, Ferdinando Nicoletti4, Jörg Bäsecke5, Sanja Mijatovic6, Danijela Maksimovic- Ivanic6, Michele Milella7, Agostino Tafuri8, Francesca Chiarini9, Camilla Evangelisti9, Lucio Cocco10, Alberto M. Martelli9,10 1 Department of Microbiology and Immunology, Brody School of Medicine at East Carolina University, Greenville, NC, USA 2 Department of Internal Medicine and Specialties, University of Palermo, Palermo, Italy 3 Consiglio Nazionale delle Ricerche, Istituto di Biomedicina e Immunologia Molecolare “Alberto Monroy”, Palermo, Italy 4 Department of Bio-Medical Sciences, University of Catania, Catania, Italy 5 Department of Medicine, University of Göttingen, Göttingen, Germany 6 Department of Immunology, Instititue for Biological Research “Sinisa Stankovic”, University of Belgrade, Belgrade, Serbia 7 Regina Elena National Cancer Institute, Rome, Italy 8 Sapienza, University of Rome, Department of Cellular Biotechnology and Hematology, Rome, Italy 9 Institute of Molecular Genetics, National Research Council-Rizzoli Orthopedic Institute, Bologna, Italy. 10 Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy Correspondence to: James A. McCubrey, email: [email protected] Keywords: Targeted Therapy, Therapy Resistance, Cancer Stem Cells, Raf, Akt, PI3K, mTOR Received: September 12, 2012, Accepted: October 18, 2012, Published: October 20, 2012 Copyright: © McCubrey et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.