The Radiochemistry of Barium, Calcium, and Strontium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Preparation of Barium Strontium Titanate Powder from Citrate
APPLIED ORGANOMETALLIC CHEMISTRY Appl. Organometal. Chem. 13, 383–397 (1999) Preparation of Barium Strontium Titanate Powder from Citrate Precursor Chen-Feng Kao* and Wein-Duo Yang Department of Chemical Engineering, National Cheng Kung University, Tainan, 70101, Taiwan TiCl4 or titanium isopropoxide reacted with INTRODUCTION citric acid to form a titanyl citrate precipitate. Barium strontium citrate solutions were then BaTiO3 is ferroelectric and piezoelectric and has added to the titanyl citrate reaction to form gels. extensive applications as an electronic material. It These gels were dried and calcined to (Ba,Sr)- can be used as a capacitor, thermistor, transducer, TiO3 powders. The gels and powders were accelerometer or degausser of colour television. characterized by DSC/TGA, IR, SEM and BaTiO3 doped with strontium retains its original XRD analyses. These results showed that, at characteristics but has a lower Curie temperature 500 °C, the gels decomposed to Ba,Sr carbonate for positive temperature coefficient devices under and TiO2, followed by the formation of (Ba,Sr)- various conditions. TiO3. The onset of perovskite formation oc- Besides solid-state reactions, chemical reactions curred at 600 °C, and was nearly complete at have also been used to prepare BaTiO3 powder. 1 1000 °C. Traces of SrCO3 were still present. Among them the hydrolysis of metal alkoxide , The cation ratios of the titanate powder oxalate precipitation in ethanol2, and alcoholic prepared in the pH range 5–6 were closest to dehydration of citrate solution3 are among the more the original stoichiometry. Only 0.1 mol% of the attractive methods. In 1956 Clabaugh et al.4 free cations remained in solution. -

PUBLIC HEALTH STATEMENT Cesium CAS#: 7440-46-2
PUBLIC HEALTH STATEMENT Cesium CAS#: 7440-46-2 Division of Toxicology April 2004 This Public Health Statement is the summary exposed to a substance when you come in contact chapter from the Toxicological Profile for cesium. It with it. You may be exposed by breathing, eating, is one in a series of Public Health Statements about or drinking the substance, or by skin contact. If the hazardous substances and their health effects. A substance is radioactive, you may also be exposed shorter version, the ToxFAQs™, is also available. to radiation if you are near it. This information is important because this substance may harm you. The effects of exposure to External exposure to radiation may occur from any hazardous substance depend on the dose, the natural or man-made sources. Naturally occurring duration, how you are exposed, personal traits and sources of radiation are cosmic radiation from space habits, and whether other chemicals are present. For or radioactive materials in soil or building materials. more information, call the ATSDR Information Man-made sources of radioactive materials are Center at 1-888-422-8737. found in consumer products, industrial equipment, _____________________________________ atom bomb fallout, and to a smaller extent from This public health statement tells you about cesium hospital waste, medical devices, and nuclear and the effects of exposure. reactors. The Environmental Protection Agency (EPA) If you are exposed to cesium, many factors identifies the most serious hazardous waste sites in determine whether you’ll be harmed. These factors the nation. These sites make up the National include the dose (how much), the duration (how Priorities List (NPL) and are the sites targeted for long), and how you come in contact with it. -

Of the Periodic Table
of the Periodic Table teacher notes Give your students a visual introduction to the families of the periodic table! This product includes eight mini- posters, one for each of the element families on the main group of the periodic table: Alkali Metals, Alkaline Earth Metals, Boron/Aluminum Group (Icosagens), Carbon Group (Crystallogens), Nitrogen Group (Pnictogens), Oxygen Group (Chalcogens), Halogens, and Noble Gases. The mini-posters give overview information about the family as well as a visual of where on the periodic table the family is located and a diagram of an atom of that family highlighting the number of valence electrons. Also included is the student packet, which is broken into the eight families and asks for specific information that students will find on the mini-posters. The students are also directed to color each family with a specific color on the blank graphic organizer at the end of their packet and they go to the fantastic interactive table at www.periodictable.com to learn even more about the elements in each family. Furthermore, there is a section for students to conduct their own research on the element of hydrogen, which does not belong to a family. When I use this activity, I print two of each mini-poster in color (pages 8 through 15 of this file), laminate them, and lay them on a big table. I have students work in partners to read about each family, one at a time, and complete that section of the student packet (pages 16 through 21 of this file). When they finish, they bring the mini-poster back to the table for another group to use. -

Barite (Barium)
Barite (Barium) Chapter D of Critical Mineral Resources of the United States—Economic and Environmental Geology and Prospects for Future Supply Professional Paper 1802–D U.S. Department of the Interior U.S. Geological Survey Periodic Table of Elements 1A 8A 1 2 hydrogen helium 1.008 2A 3A 4A 5A 6A 7A 4.003 3 4 5 6 7 8 9 10 lithium beryllium boron carbon nitrogen oxygen fluorine neon 6.94 9.012 10.81 12.01 14.01 16.00 19.00 20.18 11 12 13 14 15 16 17 18 sodium magnesium aluminum silicon phosphorus sulfur chlorine argon 22.99 24.31 3B 4B 5B 6B 7B 8B 11B 12B 26.98 28.09 30.97 32.06 35.45 39.95 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 potassium calcium scandium titanium vanadium chromium manganese iron cobalt nickel copper zinc gallium germanium arsenic selenium bromine krypton 39.10 40.08 44.96 47.88 50.94 52.00 54.94 55.85 58.93 58.69 63.55 65.39 69.72 72.64 74.92 78.96 79.90 83.79 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 rubidium strontium yttrium zirconium niobium molybdenum technetium ruthenium rhodium palladium silver cadmium indium tin antimony tellurium iodine xenon 85.47 87.62 88.91 91.22 92.91 95.96 (98) 101.1 102.9 106.4 107.9 112.4 114.8 118.7 121.8 127.6 126.9 131.3 55 56 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 cesium barium hafnium tantalum tungsten rhenium osmium iridium platinum gold mercury thallium lead bismuth polonium astatine radon 132.9 137.3 178.5 180.9 183.9 186.2 190.2 192.2 195.1 197.0 200.5 204.4 207.2 209.0 (209) (210)(222) 87 88 104 105 106 107 108 109 110 111 112 113 114 115 116 -

Paediatric Barium Swallow/Meal Information for Parents and Carers Your Doctor Has Referred Your Child for a Barium Swallow/Meal to Look at Their Food Pipe and Stomach
Oxford University Hospitals NHS Trust Paediatric Barium Swallow/Meal Information for parents and carers Your doctor has referred your child for a barium swallow/meal to look at their food pipe and stomach. We have written this information leaflet to help you and your child to understand this test. We hope it answers some of the questions you both may have and helps you and your child to feel more prepared and relaxed. page 2 What is a barium swallow/meal? A barium swallow/meal uses a special liquid to allow the X-ray doctor (radiologist) to see your child’s oesophagus (food pipe or gullet), stomach and the first part of their intestines using an X-ray camera. What does this test involve? Your child will be asked to drink a special drink called barium. This is a white liquid with a chalky texture. It is not radioactive but is used as it shows up well on X-ray images. Your child will need to drink the barium whilst lying on a couch with the X-ray camera over their tummy (about 75cm away). They may need to change position, so you will be asked to stay close to them at all times to make sure they are safe. If your child is able to, they will usually be asked to drink using a straw so that they can stay lying down. If they are not able to use a straw they can drink the barium from a bottle or beaker. If they are currently being fed through a nasogastric (NG) tube, we will use that to give them the barium. -

Absorption of X-Rays by Lead Glasses and Lead Barium Glasses
U. S. DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS RESEARCH PAPER RP870 Part of Journal of Research of the National Bureau of Standards, Volume 16, March 1936 ABSORPTION OF X"RAYS BY LEAD GLASSES AND LEAD BARIUM GLASSES By George Singer ABSTRACT The results of a study of the protective properties of a group of typical flint and barium-flint glasses are reported. In chemical composition, the protective glasses analyzed were found to resemble closely the denser optical flint and barium-flint glasses. The protection coefficients of the glasses were determined by an ionization method; of the various component elements it was found that only lead and barium contribute appreciably to the protective effectiveness of the glasses. For flint glass empirical relations were established between the protection coefficient and the chemical composition, density, and refractivity; for barium flint glass an empirical relation is given between the protection coefficient and the lead-oxide and barium-oxide components of the glass. CONTENTS Page 1. Introduction__ __ _ _ _ _ __ _ _ _ __ _ _ _ __ _ _ _ __ _ _ _ _ _ _ _ ___ _ _ _ _ _ _ _ _ _ _ _ _ _ 233 II. Methods of determining the protection coefficient of a materiaL___ 234 III. Experimental procedure_ ___ _ __ _ _ _ _ _ _ __ __ _ _ _ _ _ _ __ _ _ _ __ _ _ _ _ _ _ _ _ 235 IV. Description of glasses_ __ _ _ _ __ _ _ _ __ _ _ __ __ __ _ _ _ ______ __ _ _ _ _ _ _ _ _ 237 V. -

208143 Barium Sulfate Statistical Prea
Table of Contents LIST OF TABLES ........................................................................................................................ 3 1. EXECUTIVE SUMMARY ................................................................................................ 4 2. INTRODUCTION............................................................................................................... 7 2.1 OVERVIEW .......................................................................................................................... 7 2.1.1 Regulatory History .......................................................................................................... 7 2.1.2 Doses ............................................................................................................................... 8 2.1.3 Identified Studies in the review ....................................................................................... 8 2.1.4 Analysis Populations ....................................................................................................... 8 2.2 DATA SOURCES ................................................................................................................ 10 3. STATISTICAL EVALUATION...................................................................................... 11 3.1 DATA AND ANALYSIS QUALITY ........................................................................................ 11 3.2 EVALUATION OF EFFICACY .............................................................................................. -

The Role of Calcium and Strontium As the Most Dominant Elements During
crystals Article The Role of Calcium and Strontium as the Most Dominant Elements during Combinations of Different Alkaline Earth Metals in the Synthesis of Crystalline Silica-Carbonate Biomorphs Mayra Cuéllar-Cruz 1,2,* and Abel Moreno 2,* 1 Departamento de Biología, División de Ciencias Naturales y Exactas, Campus Guanajuato, Universidad de Guanajuato, Noria Alta S/N, Col. Noria Alta, Guanajuato C.P. 36050, Mexico 2 Instituto de Química, Universidad Nacional Autónoma de México, Av. Universidad 3000, Ciudad Universitaria, Ciudad de México 04510, Mexico * Correspondence: [email protected] (M.C.-C.); [email protected] (A.M.) Received: 22 June 2019; Accepted: 22 July 2019; Published: 24 July 2019 Abstract: The origin of life from the chemical point of view is an intriguing and fascinating topic, and is of continuous interest. Currently, the chemical elements that are part of the different cellular types from microorganisms to higher organisms have been described. However, although science has advanced in this context, it has not been elucidated yet which were the first chemical elements that gave origin to the first primitive cells, nor how evolution eliminated or incorporated other chemical elements to give origin to other types of cells through evolution. Calcium, barium, and strontium silica-carbonates have been obtained in vitro and named biomorphs, because they mimic living organism structures. Therefore, it is considered that these forms can resemble the first structures that were part of primitive organisms. Hence, the objective of this work was to synthesize biomorphs starting with different mixtures of alkaline earth metals—beryllium (Be2+), magnesium (Mg2+), calcium (Ca2+), barium (Ba2+), and strontium (Sr2+)—in the presence of nucleic acids, RNA and genomic DNA (gDNA). -
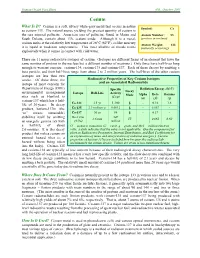
Cesium What Is It? Cesium Is a Soft, Silvery White-Gray Metal That Occurs in Nature Symbol: Cs As Cesium-133
Human Health Fact Sheet ANL, October 2001 Cesium What Is It? Cesium is a soft, silvery white-gray metal that occurs in nature Symbol: Cs as cesium-133. The natural source yielding the greatest quantity of cesium is the rare mineral pollucite. American ores of pollucite, found in Maine and Atomic Number: 55 South Dakota, contain about 13% cesium oxide. Although it is a metal, (protons in nucleus) cesium melts at the relatively low temperature of 28o C (82o F), so like mercury Atomic Weight: 133 it is liquid at moderate temperatures. This most alkaline of metals reacts (naturally occurring) explosively when it comes in contact with cold water. There are 11 major radioactive isotopes of cesium. (Isotopes are different forms of an element that have the same number of protons in the nucleus but a different number of neutrons.) Only three have half-lives long enough to warrant concern: cesium-134, cesium-135 and cesium-137. Each of these decays by emitting a beta particle, and their half-lives range from about 2 to 2 million years. The half-lives of the other cesium isotopes are less than two weeks. Of these three, the Radioactive Properties of Key Cesium Isotopes isotope of most concern for and an Associated Radionuclide Department of Energy (DOE) Specific Radiation Energy (MeV) Decay environmental management Isotope Half-Life Activity Mode Alpha Beta Gamma sites such as Hanford is (Ci/g) (α) (β) (γ) cesium-137 which has a half- 2.1 yr 1,300 β - 0.16 1.6 life of 30 years. Its decay Cs-134 product, barium-137m (the Cs-135 2.3 million yr 0.0012 β - 0.067 - “m” means metastable) Cs-137 30 yr 88 β - 0.19 - stabilizes itself by emitting Ba-137m 540 2.6 min IT - 0.065 0.60 an energetic gamma ray with (95%) million a half-life of about IT = isomeric transition, Ci = curie, g = gram, and MeV = million electron 2.6 minutes. -

Reactive Metals Hazards Packaging
Document No: RXM20172301 Publication Date: January 23, 2017 Revised Date: October 25, 2017 Hazard Awareness & Packaging Guidelines for Reactive Metals General Due to recent events resulting from reactive metals handling, CEI personnel and clients are being updated regarding special packaging guidelines designed to protect the safety of our personnel, physical assets, and customer environments. CEI’s Materials Management staff, in conjunction with guidelines from third party disposal outlets, has approved these alternative packaging guidelines to provide for safe storage and transportation of affected materials. This protocol primarily impacts water reactive or potentially water reactive metals in elemental form, although there are many compounds that are also affected. The alkali metals are a group in the periodic table consisting of the chemical elements lithium, sodium , potassium, rubidium, cesium and francium. This group lies in the s-block of the periodic table as all alkali metals have their outermost electron in an s-orbital. The alkali metals provide the best example of group trends in properties in the periodic table, with elements exhibiting well- characterized homologous behavior. The alkali metals have very similar properties: they are all shiny, soft, highly reactive metals at standard temperature and pressure, and readily lose their outermost electron to form cations with charge +1. They can all be cut easily with a knife due to their softness, exposing a shiny surface that tarnishes rapidly in air due to oxidation. Because of their high reactivity, they must be stored under oil to prevent reaction with air, and are found naturally only in salts and never as the free element. -

The Elements.Pdf
A Periodic Table of the Elements at Los Alamos National Laboratory Los Alamos National Laboratory's Chemistry Division Presents Periodic Table of the Elements A Resource for Elementary, Middle School, and High School Students Click an element for more information: Group** Period 1 18 IA VIIIA 1A 8A 1 2 13 14 15 16 17 2 1 H IIA IIIA IVA VA VIAVIIA He 1.008 2A 3A 4A 5A 6A 7A 4.003 3 4 5 6 7 8 9 10 2 Li Be B C N O F Ne 6.941 9.012 10.81 12.01 14.01 16.00 19.00 20.18 11 12 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 3 Na Mg IIIB IVB VB VIB VIIB ------- VIII IB IIB Al Si P S Cl Ar 22.99 24.31 3B 4B 5B 6B 7B ------- 1B 2B 26.98 28.09 30.97 32.07 35.45 39.95 ------- 8 ------- 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 4 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 39.10 40.08 44.96 47.88 50.94 52.00 54.94 55.85 58.47 58.69 63.55 65.39 69.72 72.59 74.92 78.96 79.90 83.80 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 5 Rb Sr Y Zr NbMo Tc Ru Rh PdAgCd In Sn Sb Te I Xe 85.47 87.62 88.91 91.22 92.91 95.94 (98) 101.1 102.9 106.4 107.9 112.4 114.8 118.7 121.8 127.6 126.9 131.3 55 56 57 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 6 Cs Ba La* Hf Ta W Re Os Ir Pt AuHg Tl Pb Bi Po At Rn 132.9 137.3 138.9 178.5 180.9 183.9 186.2 190.2 190.2 195.1 197.0 200.5 204.4 207.2 209.0 (210) (210) (222) 87 88 89 104 105 106 107 108 109 110 111 112 114 116 118 7 Fr Ra Ac~RfDb Sg Bh Hs Mt --- --- --- --- --- --- (223) (226) (227) (257) (260) (263) (262) (265) (266) () () () () () () http://pearl1.lanl.gov/periodic/ (1 of 3) [5/17/2001 4:06:20 PM] A Periodic Table of the Elements at Los Alamos National Laboratory 58 59 60 61 62 63 64 65 66 67 68 69 70 71 Lanthanide Series* Ce Pr NdPmSm Eu Gd TbDyHo Er TmYbLu 140.1 140.9 144.2 (147) 150.4 152.0 157.3 158.9 162.5 164.9 167.3 168.9 173.0 175.0 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Actinide Series~ Th Pa U Np Pu AmCmBk Cf Es FmMdNo Lr 232.0 (231) (238) (237) (242) (243) (247) (247) (249) (254) (253) (256) (254) (257) ** Groups are noted by 3 notation conventions. -

Compounds of the Metals Beryllium, Magnesium
C01F COMPOUNDS OF THE METALS BERYLLIUM, MAGNESIUM, ALUMINIUM, CALCIUM, STRONTIUM, BARIUM, RADIUM, THORIUM, OR OF THE RARE-EARTH METALS (metal hydrides [N: monoborane, diborane or addition complexes thereof] C01B6/00; salts of oxyacids of halogens C01B11/00; peroxides, salts of peroxyacids C01B15/00; sulfides or polysulfides of magnesium, calcium, strontium, or barium C01B17/42; thiosulfates, dithionites, polythionates C01B17/64; compounds containing selenium or tellurium C01B19/00; binary compounds of nitrogen with metals C01B21/06; azides C01B21/08; [N: compounds other than ammonia or cyanogen containing nitrogen and non-metals and optionally metals C01B21/082; amides or imides of silicon C01B21/087]; metal [N: imides or] amides C01B21/092, [N: C01B21/0923]; nitrites C01B21/50; [N: compounds of noble gases C01B23/0005]; phosphides C01B25/08; salts of oxyacids of phosphorus C01B25/16; carbides C01B31/30; compounds containing silicon C01B33/00; compounds containing boron C01B35/00; compounds having molecular sieve properties but not having base-exchange properties C01B37/00; compounds having molecular sieve and base-exchange properties, e.g. crystalline zeolites, C01B39/00;cyanides C01C3/08; salts of cyanic acid C01C3/14; salts of cyanamide C01C3/16; thiocyanates C01C3/20; [N: double sulfates of magnesium with sodium or potassium C01D5/12; with other alkali metals C01D15/00, C01D17/00]) Definition statement This subclass/group covers: All compounds of Be,Mg,Al,Ca,Sr,Ba,Ra,Th or rare earth metals except those compounds which are classified in C01G because of application of the last appropriate place rule. So, in principle does this subclass comprise all Al-compounds with elements as such being part of C01B-C01D, e.g.