Encephalopathy 1 5 Steven L
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chasing the Dragon, a Case of Leukoencephalopathy Associated with Heroin Inhalation
Chasing the dragon, a case of leukoencephalopathy associated with heroin inhalation Daniel Cho MD1, Hani Nazha MD2, Kalin Fisher BS2 Author Affiliations: 1. Charleston Area Medical Center, Charleston, West Virginia 2. West Virginia University, Morgantown, West Virginia The authors have no financial disclosures to declare and no conflicts of interest to report. Corresponding Author: Hani Nazha MD West Virginia University Morgantown, West Virginia Email: [email protected] Abstract Although rare, toxic leukoencephalopathy (TLE) associated with heroin inhalation has been reported. “Chasing the dragon” may lead to progressive spongiform degeneration of the brain and presents with a large range of neuropsychological sequelae. This case is an example of TLE in a middle-aged white male with a history of polysubstance abuse. He presented with a three week history of progressive neuropsychological symptoms, including abulia, bradyphrenia, hyperreflexia, and visual hallucinations. He was initially suspected to have progressive multifocal leukoencephalopathy, however, JCV PCR was negative. MRI showed diffuse abnormal signal in the white matter, extending into the thalami and cerebral peduncles. Brain biopsy was performed, which revealed spongiform degeneration, and a diagnosis of TLE was made. The patient was then transferred to a skilled nursing facility. Clinical suspicion based on a thorough history and clinical exam findings is paramount in recognition of heroin-associated TLE. Although rare, heroin-inhalation TLE continues to be reported. As ‘chasing the dragon’ is gaining popularity among drug users, it is important for clinicians to be able to recognize this disease process. Keywords Opioid, Addiction, Heroin, Leukoencephalopathy Introduction Toxic leukoencephalopathy (TLE) associated with heroin abuse was first described in 1982.1 “Chasing the dragon" is a method of heroin vapor inhalation in which a small amount of heroin powder is placed on aluminum foil, which is then heated by placing a match or lighter underneath. -

Para-Infectious and Post-Vaccinal Encephalomyelitis
Postgrad Med J: first published as 10.1136/pgmj.45.524.392 on 1 June 1969. Downloaded from Postgrad. med. J. (June 1969) 45, 392-400. Para-infectious and post-vaccinal encephalomyelitis P. B. CROFT The Tottenham Group ofHospitals, London, N.15 Summary of neurological disorders following vaccinations of The incidence of encephalomyelitis in association all kinds (de Vries, 1960). with acute specific fevers and prophylactic inocula- The incidence of para-infectious and post-vaccinal tions is discussed. Available statistics are inaccurate, encephalomyelitis in Great Britain is difficult to but these conditions are of considerable importance estimate. It is certain that many cases are not -it is likely that there are about 400 cases ofmeasles notified to the Registrar General, whose published encephalitis in England and Wales in an epidemic figures must be an underestimate. In addition there year. is a lack of precise diagnostic criteria and this aspect The pathology of these neurological complications will be considered later. In the years 1964-66 the is discussed and emphasis placed on the distinction mean number of deaths registered annually in between typical perivenous demyelinating encepha- England and Wales as due to acute infectious litis, and the toxic type of encephalopathy which encephalitis was ninety-seven (Registrar General, occurs mainly in young children. 1967). During the same period the mean annual The clinical syndromes occurring in association number of deaths registered as due to the late effects with measles, chickenpox and German measles are of acute infectious encephalitis was seventy-four- considered. Although encephalitis is the most fre- this presumably includes patients with post-encepha-copyright. -

MRI Brain in Monohalomethane Toxic Encephalopathy
Published online: 2021-07-30 NEURORADIOLOOGY MRI brain in monohalomethane toxic encephalopathy: A case report Yogeshwari S Deshmukh, Ashish Atre, Darshan Shah, Sudhir Kothari1 Departments of Radiology, Star Imaging and Research Centre, Opp. Kamala Nehru Park, Joshi Hospital Campus, Erandwane, 1Neurology OPD, Poona Hospital and Research Centre, 27, Sadashiv Peth, Pune, Maharashtra, India Correspondence: Dr. Yogeshwari S. Deshmukh, Department of Radiology, Star Imaging and Research Centre, Opp. Kamala Nehru Park, Joshi Hospital Campus, Erandwane, Pune - 411 004, Maharashtra, India. E-mail: [email protected] Abstract Monohalomethanes are alkylating agents that have been used as methylating agents, laboratory reagents, refrigerants, aerosol propellants, pesticides, fumigants, fire‑extinguishing agents, anesthetics, degreasers, blowing agents for plastic foams, and chemical intermediates. Compounds in this group are methyl chloride, methyl bromide, methyl iodide (MI), and methyl fluoride. MI is a colorless volatile liquid used as a methylating agent to manufacture a few pharmaceuticals and is also used as a fumigative insecticide. It is a rare intoxicant. Neurotoxicity is known with both acute and chronic exposure to MI. We present the characteristic magnetic resonance imaging (MRI) brain findings in a patient who developed neuropsychiatric symptoms weeks after occupational exposure to excessive doses of MI. Key words: Methyl iodide; MRI brain; toxic encephalopathy Introduction can cause cranial neuropathy, pyramidal and cerebellar dysfunction, polyneuropathies, and behavioral changes. Methyl iodide (MI) is a monohalothane used as an organic chemical agent/methylating agent in the synthesis Few case reports have been described, however.[1‑5] As far of various pharmaceutical drugs. It is also used as a as we know, none of them mention abnormal magnetic fumigative pesticide commonly in greenhouses and resonance imaging (MRI) findings except one.[1] strawberry farming. -

Encephalopathy and Encephalitis Associated with Cerebrospinal
SYNOPSIS Encephalopathy and Encephalitis Associated with Cerebrospinal Fluid Cytokine Alterations and Coronavirus Disease, Atlanta, Georgia, USA, 2020 Karima Benameur,1 Ankita Agarwal,1 Sara C. Auld, Matthew P. Butters, Andrew S. Webster, Tugba Ozturk, J. Christina Howell, Leda C. Bassit, Alvaro Velasquez, Raymond F. Schinazi, Mark E. Mullins, William T. Hu There are few detailed investigations of neurologic unnecessary staff exposure and difficulties in estab- complications in severe acute respiratory syndrome lishing preillness neurologic status without regular coronavirus 2 infection. We describe 3 patients with family visitors. It is known that neurons and glia ex- laboratory-confirmed coronavirus disease who had en- press the putative SARS-CoV-2 receptor angiotensin cephalopathy and encephalitis develop. Neuroimaging converting enzyme 2 (7), and that the related coro- showed nonenhancing unilateral, bilateral, and midline navirus SARS-CoV (responsible for the 2003 SARS changes not readily attributable to vascular causes. All 3 outbreak) can inoculate the mouse olfactory bulb (8). patients had increased cerebrospinal fluid (CSF) levels If SARS-CoV-2 can enter the central nervous system of anti-S1 IgM. One patient who died also had increased (CNS) directly or through hematogenous spread, ce- levels of anti-envelope protein IgM. CSF analysis also rebrospinal fluid (CSF) changes, including viral RNA, showed markedly increased levels of interleukin (IL)-6, IgM, or cytokine levels, might support CNS infec- IL-8, and IL-10, but severe acute respiratory syndrome coronavirus 2 was not identified in any CSF sample. tion as a cause for neurologic symptoms. We report These changes provide evidence of CSF periinfectious/ clinical, blood, neuroimaging, and CSF findings for postinfectious inflammatory changes during coronavirus 3 patients with laboratory-confirmed COVID-19 and disease with neurologic complications. -

Neurological Disorders in Liver Transplant Candidates: Pathophysiology ☆ and Clinical Assessment
Transplantation Reviews 31 (2017) 193–206 Contents lists available at ScienceDirect Transplantation Reviews journal homepage: www.elsevier.com/locate/trre Neurological disorders in liver transplant candidates: Pathophysiology ☆ and clinical assessment Paolo Feltracco a,⁎, Annachiara Cagnin b, Cristiana Carollo a, Stefania Barbieri a, Carlo Ori a a Department of Medicine UO Anesthesia and Intensive Care, Padova University Hospital, Padova, Italy b Department of Neurosciences (DNS), University of Padova, Padova, Italy abstract Compromised liver function, as a consequence of acute liver insufficiency or severe chronic liver disease may be associated with various neurological syndromes, which involve both central and peripheral nervous system. Acute and severe hyperammoniemia inducing cellular metabolic alterations, prolonged state of “neuroinflamma- tion”, activation of brain microglia, accumulation of manganese and ammonia, and systemic inflammation are the main causative factors of brain damage in liver failure. The most widely recognized neurological complications of serious hepatocellular failure include hepatic encephalopathy, diffuse cerebral edema, Wilson disease, hepatic myelopathy, acquired hepatocerebral degeneration, cirrhosis-related Parkinsonism and osmotic demyelination syndrome. Neurological disorders affecting liver transplant candidates while in the waiting list may not only sig- nificantly influence preoperative morbidity and even mortality, but also represent important predictive factors for post-transplant neurological manifestations. -

TDP-43 Proteinopathy and Motor Neuron Disease in Chronic Traumatic Encephalopathy
J Neuropathol Exp Neurol Vol. 69, No. 9 Copyright Ó 2010 by the American Association of Neuropathologists, Inc. September 2010 pp. 918Y929 ORIGINAL ARTICLE TDP-43 Proteinopathy and Motor Neuron Disease in Chronic Traumatic Encephalopathy Ann C. McKee, MD, Brandon E. Gavett, PhD, Robert A. Stern, PhD, Christopher J. Nowinski, AB, Robert C. Cantu, MD, Neil W. Kowall, MD, Daniel P. Perl, MD, E. Tessa Hedley-Whyte, MD, Bruce Price, MD, Chris Sullivan, Peter Morin, MD, PhD, Hyo-Soon Lee, MD, Caroline A. Kubilus, Daniel H. Daneshvar, MA, Megan Wulff, MPH, and Andrew E. Budson, MD cord in addition to tau neurofibrillary changes, motor neuron loss, Abstract and corticospinal tract degeneration. The TDP-43 proteinopathy Epidemiological evidence suggests that the incidence of amyo- associated with CTE is similar to that found in frontotemporal lobar trophic lateral sclerosis is increased in association with head injury. degeneration with TDP-43 inclusions, in that widespread regions of Repetitive head injury is also associated with the development of the brain are affected. Akin to frontotemporal lobar degeneration chronic traumatic encephalopathy (CTE), a tauopathy characterized with TDP-43 inclusions, in some individuals with CTE, the TDP-43 by neurofibrillary tangles throughout the brain in the relative absence proteinopathy extends to involve the spinal cord and is associated of A-amyloid deposits. We examined 12 cases of CTE and, in 10, with motor neuron disease. This is the first pathological evidence that found a widespread TAR DNA-binding protein of approximately repetitive head trauma experienced in collision sports might be 43 kd (TDP-43) proteinopathy affecting the frontal and temporal associated with the development of a motor neuron disease. -

Encephalopathy
Jounal ofNeurology, Neurosurgery, and Psychiatry 1997;62:51-60 51 Operational criteria for the classification of chronic alcoholics: identification of Wemicke's encephalopathy D Caine, G M Halliday, J J Kril, C G Harper Abstract suggest that there can be a complete resolution Objectives-To establish better opera- of the underlying brain abnormality in tional criteria for the diagnosis of Wernicke's encephalopathy, similar to the res- Wernicke's encephalopathy. Current olution of neuropathology in hepatic criteria for diagnosing Wernicke's encephalopathy. However, unlike hepatic encephalopathy require the presence of encephalopathy, in which the clinical signs are three clinical signs (oculomotor abnor- a precursor of the pathology, many patients malities, cerebellar dysfunction, and an with the neuropathology of Wernicke's altered mental state), although it has encephalopathy do not have recorded signs of often been reported that most patients do the classic triad.' 2 This raises the question of not filfil all these criteria. whether the clinical correlates of the patholog- Methods-The clinical histories of 28 ical lesions are being missed during life or alcoholics with neurological and neu- whether the pathology represents clinically ropsychological assessments and defini- silent lesions. A similar situation is seen in tive neuropathological diagnoses were Alzheimer's disease, in which many non- examined to determine clinical signs for demented aged patients show a similar type use in a screening schedule. Operational and distribution of neuropathology to their criteria were then proposed for differenti- demented counterparts. 12 Recently research ating patients with Wernicke's into the pathophysiology of aging and encephalopathy alone or in combination Alzheimer's disease has been successfully with Korsakoff's psychosis or hepatic advanced by the use of operational criteria encephalopathy. -

Acute Encephalopathy with Bilateral Thalamotegmental Involvement in Infants and Children: Imaging and Pathology Findings
Acute Encephalopathy with Bilateral Thalamotegmental Involvement in Infants and Children: Imaging and Pathology Findings Akira Yagishita, Imaharu Nakano, Takakazu Ushioda, Noriyuki Otsuki, and Akio Hasegawa PURPOSE: To investigate the imaging and pathologic characteristics of acute encephalopathy with bilateral thalamotegmental involvement in infants and children. METHODS: Five Japanese children ranging in age from 11 to 29 months were studied. We performed CT imaging in all patients, 10 MR examinations in four patients, and an autopsy in one patient. RESULTS: The encephalopathy affected the thalami, brain stem tegmenta, and cerebral and cerebellar white matter. The brain of the autopsied case showed fresh necrosis and brain edema without inflam- matory cell infiltration. Petechiae and congestion were demonstrated mainly in the thalamus. CT and MR images showed symmetric focal lesions in the same areas in the early phase. These lesions became more demarcated and smaller in the intermediate phase. The ventricles and cortical sulci enlarged. MR images demonstrated T1 shortening in the thalami. The prognosis was generally poor; one patient died, three patients were left with severe sequelae, and only one patient im- proved. CONCLUSIONS: The encephalopathy might be a postviral or postinfectious brain disor- der. T1 shortening in the thalami indicated the presence of petechiae. Index terms: Brain, diseases; Brain, magnetic resonance; Infants, diseases AJNR Am J Neuroradiol 16:439–447, March 1995 A strange type of acute encephalopathy has and cerebellar white matter. All patients in been reported in Japan (1–12). The acute en- whom this condition has been diagnosed thus cephalopathy occurs in infants and young chil- far have been Japanese infants and children, as dren without sexual predilection and is pre- far as we know. -

Acute Leucoencephalopathy with Restriction of Diffusion-A Case Report
Eastern Journal of Medicine 17 (2012) 149-152 V. Singh et al / Dwi in acute leucoencephalopathy Case Report Acute leucoencephalopathy with restriction of diffusion-a case report Vivek Singh, Vaishali Tomar, Alok Kumar, Rajendra V. Phadke* Department of Radiodiagnosis, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, India Abstract. Acute white matter insult in a non traumatic, non ischemic background has been recently identified in certain clinical situations. Named toxic leukoencephalopathy, it may be caused by exposure to a wide variety of agents, including cranial irradiation, therapeutic agents, drugs of abuse, and environmental toxins. Extensive central white matter restriction of diffusion in the absence of significant T2 or Fluid-attenuated inversion recovery (FLAIR) abnormality is the imaging hallmark on Magnetic Resonance Imaging (MRI). Reversibility of the symptoms after the withdrawal of toxin has been shown in a few reports. We present one such case with reversal of changes on MRI and clinical improvement. Key words: ADEM, intramyelinic edema, PRES, toxic leukoencephalopathy 1. Introduction 2. Case report Leukoencephalopathy is a structural alteration A 7 year-old male child presented elsewhere of cerebral white matter in which myelin suffers with a short history of mild fever and had two the most damage (1). Toxic leukoencephalopathy episodes of vomiting. He received multiple is a recently identified entity seen in patients medications including antibiotics empirically. He exposed to a wide variety of agents like cranial went into altered sensorium on the 3rd day. There irradiation, therapeutic agents, drugs of abuse and was no history of seizures or past history of any environmental toxins (1). -

Pharmacologic Treatment of Meningitis and Encephalitis in Adult Patients
Published online: 2019-06-19 THIEME Review Article 145 Pharmacologic Treatment of Meningitis and Encephalitis in Adult Patients Andrew K. Treister1 Ines P. Koerner2 1Department of Neurology, Oregon Health & Science University, Address for correspondence Andrew K. Treister, MD, Department Portland, Oregon, United States of Neurology, Oregon Health & Science University, 3181 SW Sam 2Department of Anesthesiology and Perioperative Medicine, Jackson Park, CR 120, Portland, OR 97239-3098, United States Oregon Health & Science University, Portland, Oregon, (e-mail: [email protected]). United States J Neuroanaesthesiol Crit Care 2019;6:145–152 Abstract Meningitis and encephalitis are two inflammatory, often infectious, disorders of the meninges and the central nervous system. Both are associated with significant morbidity and mortality, and require early and aggressive targeted treatment. This article reviews pharmacologic treatment strategies for infectious meningitis and encephalitis, using Keywords the latest available research and guidelines. It provides an overview of empiric anti- ► meningitis microbial treatment approaches for a variety of organisms, including a discussion of ► encephalitis trends in antibiotic resistance where applicable. Key steps in diagnosis and general ► antibiotic resistance management are briefly reviewed. Introduction care–associated infections are not covered in detail, as they are excellently discussed in a recent guideline statement.4 The terms meningitis and encephalitis comprise a broad array of infectious and inflammatory processes involving the central nervous system (CNS) that carry significant morbidity Literature Review 1,2 and mortality. Meningitis refers to inflammation of PubMed was searched in January 2019 for articles published primarily the meninges, although it may spread to involve the between January 1, 2009, and December 31, 2018. -

Idiopathic Intracranial Hypertension
IDIOPATHIC INTRACRANIAL HYPERTENSION William L Hills, MD Neuro-ophthalmology Oregon Neurology Associates Affiliated Assistant Professor Ophthalmology and Neurology Casey Eye Institute, OHSU No disclosures CASE - 19 YO WOMAN WITH HEADACHES X 3 MONTHS Headaches frontal PMHx: obesity Worse lying down Meds: takes ibuprofen for headaches Wake from sleep Pulsatile tinnitus x 1 month. Vision blacks out transiently when she bends over or sits down EXAMINATION Vision: 20/20 R eye, 20/25 L eye. Neuro: PERRL, no APD, EOMI, VF full to confrontation. Dilated fundoscopic exam: 360 degree blurring of disc margins in both eyes, absent SVP. Formal visual field testing: Enlargement of the blind spot, generalized constriction both eyes. MRI brain: Lumbar puncture: Posterior flattening of Opening pressure 39 the globes cm H20 Empty sella Normal CSF studies otherwise normal Headache improved after LP IDIOPATHIC INTRACRANIAL HYPERTENSION SYNDROME: Increased intracranial pressure without ventriculomegaly or mass lesion Normal CSF composition NOMENCLATURE Idiopathic intracranial hypertension (IIH) Benign intracranial hypertension Pseudotumor cerebri Intracranial hypertension secondary to… DIAGNOSTIC CRITERIA Original criteria have been updated to reflect new imaging modalities: 1492 Friedman and Jacobsen. Neurology 2002; 59: Symptoms and signs reflect only those of - increased ICP or papilledema 1495 Documented increased ICP during LP in lateral decubitus position Normal CSF composition No evidence of mass, hydrocephalus, structural -
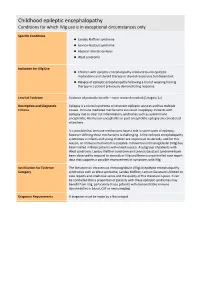
Childhood Epileptic Encephalopathy Conditions for Which Ivig Use Is in Exceptional Circumstances Only
Childhood epileptic encephalopathy Conditions for which IVIg use is in exceptional circumstances only Specific Conditions Landau Kleffner syndrome Lennox‐Gastaut syndrome Atypical rolandic epilepsy West syndrome Indication for IVIg Use Children with epileptic encephalopathy resistant to anti‐epileptic medications and steroid therapy or steroid responsive but dependant Relapse of epileptic encephalopathy following a trial of weaning from Ig therapy in a patient previously demonstrating response Level of Evidence Evidence of probable benefit – more research needed (Category 2a) Description and Diagnostic Epilepsy is a clinical syndrome of recurrent epileptic seizures and has multiple Criteria causes. Immune mediated mechanisms can result in epilepsy. Patients with epilepsy due to clear cut inflammatory syndromes such as autoimmune encephalitis, Rasmussen encephalitis or post encephalitic epilepsy are considered elsewhere. It is possible that immune mechanisms have a role in some cases of epilepsy, however defining these mechanisms is challenging. A few epileptic encephalopathy syndromes in infants and young children are responsive to steroids, and for this reason, an immune mechanism is possible. Intravenous immunoglobulin (IVIg) has been trialled in these patients with mixed success. A subgroup of patients with West syndrome, Landau Kleffner syndrome and Lennox Gaustaut syndrome have been observed to respond to steroids or IVIg and there is uncontrolled case report data that supports a possible improvement of symptoms with IVIg. Justification for Evidence The literature on intravenous immunoglobulin (IVIg) in epileptic encephalopathy Category syndromes such as West syndrome, Landau Kleffner, Lennox Gaustaut is limited to case reports and small case series and the quality of this literature is poor. It can be concluded that a proportion of patients with these epileptic syndromes may benefit from IVIg, particularly those patients with demonstrable immune abnormalities in blood, CSF or neuroimaging.