Physiology of Venous Return?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cardiac Physiologyc As a Country
Type to enter text CardiacCardiovascular PhysiologyC as a Physiology Country Doc EpisodeEpisode 2:2: The EKG Electrocardiography I Patrick Eggena, M.D. Novateur Medmedia Type EPISODEto enter text 2 Type to enter text THE EKG by Patrick Eggena, M.D. i Copyright This Episode is derived from: Course in Cardiovascular Physiology by Patrick Eggena, M.D. © Copyright Novateur Medmedia, LLC. April 13, 2012 The United States Copyright Registration Number: PAu3-662-048 Ordering Information via iBooks: ISBN 978-0-9905771-1-9 Cardiac Physiology as a Country Doc, Episode 2: The EKG. Contact Information: Novateur Medmedia, LLC 39 Terry Hill Road, Carmel, NY 10512 email: [email protected] Credits: Oil Paintings by Bonnie Eggena, PsD. Music by Alan Goodman from his CD Under the Bed, Cancoll Music, copyright 2005 (with permission). Illustrations, movies, text, and lectures by Patrick Eggena, M.D. Note: Knowledge in the basic and clinical sciences is constantly changing. The reader is advised to carefully consult the instruc- tions and informational material included in the package inserts of each drug or therapeutic agent before administration. The Country Doctor Series illustrates Physiological Principles and is not intended as a guide to Medical Therapeutics. Care has been taken to present correct information in this book, however, the author and publisher are not responsible for errors or omissions or for any consequence from application of the information in this work and make no warranty, expressed or implied, with re- spect to the contents of this publication or that its operation will be uninterrupted and error free on any particular recording de- vice. -

Fluid Resuscitation Therapy for Hemorrhagic Shock
CLINICAL CARE Fluid Resuscitation Therapy for Hemorrhagic Shock Joseph R. Spaniol vides a review of the 4 types of shock, the 4 classes of Amanda R. Knight, BA hemorrhagic shock, and the latest research on resuscita- tive fluid. The 4 types of shock are categorized into dis- Jessica L. Zebley, MS, RN tributive, obstructive, cardiogenic, and hemorrhagic Dawn Anderson, MS, RN shock. Hemorrhagic shock has been categorized into 4 Janet D. Pierce, DSN, ARNP, CCRN classes, and based on these classes, appropriate treatment can be planned. Crystalloids, colloids, dopamine, and blood products are all considered resuscitative fluid treat- ment options. Each individual case requires various resus- ■ ABSTRACT citative actions with different fluids. Healthcare Hemorrhagic shock is a severe life-threatening emergency professionals who are knowledgeable of the information affecting all organ systems of the body by depriving tissue in this review would be better prepared for patients who of sufficient oxygen and nutrients by decreasing cardiac are admitted with hemorrhagic shock, thus providing output. This article is a short review of the different types optimal care. of shock, followed by information specifically referring to hemorrhagic shock. The American College of Surgeons ■ DISTRIBUTIVE SHOCK categorized shock into 4 classes: (1) distributive; (2) Distributive shock is composed of 3 separate categories obstructive; (3) cardiogenic; and (4) hemorrhagic. based on their clinical outcome. Distributive shock can be Similarly, the classes of hemorrhagic shock are grouped categorized into (1) septic; (2) anaphylactic; and (3) neu- by signs and symptoms, amount of blood loss, and the rogenic shock. type of fluid replacement. This updated review is helpful to trauma nurses in understanding the various clinical Septic shock aspects of shock and the current recommendations for In accordance with the American College of Chest fluid resuscitation therapy following hemorrhagic shock. -

Insights from Computational Modeling Into the Contribution of Mechano-Calcium Feedback on the Cardiac End-Systolic Force-Length Relationship
fphys-11-00587 May 28, 2020 Time: 19:46 # 1 ORIGINAL RESEARCH published: 29 May 2020 doi: 10.3389/fphys.2020.00587 Insights From Computational Modeling Into the Contribution of Mechano-Calcium Feedback on the Cardiac End-Systolic Force-Length Relationship Megan E. Guidry1, David P. Nickerson1, Edmund J. Crampin2,3, Martyn P. Nash1,4, Denis S. Loiselle1,5 and Kenneth Tran1* 1 Auckland Bioengineering Institute, The University of Auckland, Auckland, New Zealand, 2 Systems Biology Laboratory, School of Mathematics and Statistics, The University of Melbourne, Melbourne, VIC, Australia, 3 ARC Centre of Excellence in Convergent Bio-Nano Science and Technology, School of Chemical and Biomedical Engineering, The University of Melbourne, Melbourne, VIC, Australia, 4 Department of Engineering Science, The University of Auckland, Auckland, Edited by: New Zealand, 5 Department of Physiology, The University of Auckland, Auckland, New Zealand Leonid Katsnelson, Institute of Immunology and Physiology (RAS), Russia In experimental studies on cardiac tissue, the end-systolic force-length relation (ESFLR) Reviewed by: has been shown to depend on the mode of contraction: isometric or isotonic. The Andrey K. Tsaturyan, Lomonosov Moscow State University, isometric ESFLR is derived from isometric contractions spanning a range of muscle Russia lengths while the isotonic ESFLR is derived from shortening contractions across a Aurore Lyon, range of afterloads. The ESFLR of isotonic contractions consistently lies below its Maastricht University, Netherlands isometric counterpart. Despite the passing of over a hundred years since the first insight *Correspondence: Kenneth Tran by Otto Frank, the mechanism(s) underlying this protocol-dependent difference in the [email protected] ESFLR remain incompletely explained. -

Integration of Detailed Modules in a Core Model of Body Fluid Homeostasis and Blood Pressure Regulation
Integration of detailed modules in a core model of body fluid homeostasis and blood pressure regulation. Alfredo Hernández, Virginie Le Rolle, David Ojeda, Pierre Baconnier, Julie Fontecave-Jallon, François Guillaud, Thibault Grosse, Robert Moss, Patrick Hannaert, Randall Thomas To cite this version: Alfredo Hernández, Virginie Le Rolle, David Ojeda, Pierre Baconnier, Julie Fontecave-Jallon, et al.. Integration of detailed modules in a core model of body fluid homeostasis and blood pres- sure regulation.. Progress in Biophysics and Molecular Biology, Elsevier, 2011, 107 (1), pp.169-82. 10.1016/j.pbiomolbio.2011.06.008. inserm-00654821 HAL Id: inserm-00654821 https://www.hal.inserm.fr/inserm-00654821 Submitted on 27 Dec 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Integration of detailed modules in a core model of body fluid homeostasis and blood pressure regulation Alfredo I. Hernández1,2, Virginie Le Rolle1,2, David Ojeda1,2, Pierre Baconnier3, Julie Fontecave-Jallon3, François Guillaud4, Thibault Grosse5,6, Robert G. Moss5,6, Patrick Hannaert4, S. Randall Thomas5,6 1INSERM, U642, Rennes, F-35000, France; 2Université de Rennes 1, LTSI, Rennes, F-35000, France; 3 UJF-Grenoble 1 / CNRS / TIMC-IMAG UMR 5525 (Equipe PRETA), Grenoble, F-38041, France 4INSERM U927. -

Regulation of the Systemic Circulation*
CIRCULATORY PHYSIOLOGY SECTION 6: REGULATION OF THE SYSTEMIC CIRCULATION* Summary: This set of notes integrates what we have learned about vascular and cardiac function into a whole so that we can see how the entire circulation is regulated. These are very important concepts and their mastery will move you a long way towards a real understanding of the operation of the circulatory system. I. The Systemic Vascular System -- The influence of cardiac output on the partition of blood between the venous and arterial sides of the systemic circulation. A. Earlier we have indicated that at rest most of the blood in the systemic circulation is located in the venous side. This is in part due to the relatively high capacitance of the venous system when compared to the arterial system. This gives a reserve of blood that can be used when demands for respiratory gas circulation increase as in exercise. B. When there is no circulation (death or experimental interruption of the cardiac output), there is still a blood pressure in both the arterial and venous systems. 1. As we indicated earlier, this is due to the elastic forces stored in the walls of the vessels -- since in the absence of a hemorrhage the vessels contain enough fluid to increase their volume beyond resting volume, there must be a pressure even if the blood is not moving. 2. This low pressure is referred to as the mean circulatory pressure (Guyton) and for normal blood volumes is about 7mmHg (know this value). 3. Due to the greater compliance of the venous system, the greatest relative proportion of the total blood volume will reside in the venous system when operating at the mean circulatory pressure (i.e., when the heart is stopped). -

Strategic Review of Cardiac Physiology Services in England: Final Report
STRATEGIC REVIEW OF CARDIAC PHYSIOLOGY SERVICES IN ENGLAND: FINAL REPORT Strategic Review of Cardiac Physiology Services in England Page 1 Final Report: 12/05/2015 CONTENTS Page Foreword 3 Summary of recommendations 4 Background 7 Purpose of the Review 8 Working arrangements and governance 8 Delivering a service model for the future 8 Delivering a cardiac physiology service to quality standards 16 Developing a cardiac physiology workforce for the future 18 Commissioning for quality 20 Appendix 1: 23 Cardiac physiology services in summary Appendix 2: 27 Working arrangements and governance Appendix 3: 28 Award in Practical Electrocardiography (APECG) Appendix 4: 29 Proposed possible key performance indicators Appendix 5: 30 Current status of standards development across a range of cardiac physiology investigations Appendix 6: 32 Current training, accreditation and standards Appendix 7: 33 National Review of Cardiac Physiology Services - Modelling the Future Workforce References 34 Glossary 36 Acknowledgements 38 Strategic Review of Cardiac Physiology Services in England Page 2 Final Report: 12/05/2015 FOREWORD In 2013, the Department of Health published the Cardiovascular Outcomes Strategy, a call to action to the health and care systems, which set out ten key actions to deliver for patients with cardiovascular disease improvements in their health outcomes. Cardiac physiology services are key to delivering the improved treatment and care, which is the ambition of the Outcomes Strategy. This Strategic Review of Cardiac Physiology Services was set up to make recommendations to NHS England, Health Education England and across the health and care system about the innovations in service delivery, models of care and workforce configuration which are needed to transform patients’ experience of cardiovascular disease care. -

Cardiac Physiology
Chapter 20 Cardiac Physiology LENA S. SUN • JOHANNA SCHWARZENBERGER • RADHIKA DINAVAHI K EY P OINTS • The cardiac cycle is the sequence of electrical and mechanical events during the course of a single heartbeat. • Cardiac output is determined by the heart rate, myocardial contractility, and preload and afterload. • The majority of cardiomyocytes consist of myofibrils, which are rodlike bundles that form the contractile elements within the cardiomyocyte. • The basic working unit of contraction is the sarcomere. • Gap junctions are responsible for the electrical coupling of small molecules between cells. • Action potentials have four phases in the heart. • The key player in cardiac excitation-contraction coupling is the ubiquitous second messenger calcium. • Calcium-induced sparks are spatially and temporally patterned activations of localized calcium release that are important for excitation-contraction coupling and regulation of automaticity and contractility. • β -Adrenoreceptors stimulate chronotropy, inotropy, lusitropy, and dromotropy. • Hormones with cardiac action can be synthesized and secreted by cardiomyocytes or produced by other tissues and delivered to the heart. • Cardiac reflexes are fast-acting reflex loops between the heart and central nervous system that contribute to the regulation of cardiac function and the maintenance of physiologic homeostasis. In 1628, English physician, William Harvey, first advanced oxygen (O2) and nutrients and to remove carbon dioxide the modern concept of circulation with the heart as the (CO2) and metabolites from various tissue beds. generator for the circulation. Modern cardiac physiology includes not only physiology of the heart as a pump but also concepts of cellular and molecular biology of the car- PHYSIOLOGY OF THE INTACT HEART diomyocyte and regulation of cardiac function by neural and humoral factors. -

Ventricular End-Diastolic Volume (Ml)
Stroke Volume and Heart Failure Black/Blue: text. Red: very important. Green: Doctor’s notes. Pink: formulas. Yellow: numbers. Gray: notes and explanation. Physiology Team 436 – Cardiovascular Block Lecture 9 Lecture: If work is intended for initial studying. Review: If work is intended for revision. 1 Objectives Study Smart: focus on mutual topics. From the students’ guide: Explain how cardiac contractility affect stroke volume. Calculate CO using Fick’s principle equation. Explain pathophysiology of heart failure and differentiate between left and right failure. Explain how the pathophysiology associated with heart failure results in typical signs and symptoms. 2 Stroke Volume Stroke volume: is the volume of blood pumped (ejected) by each ventricle per beat (during each ventricular systole), and it is about 70-80 ml/beat. Factors Affecting It: 1- End diastolic volume (EDV) (Preload): • It is: the volume of blood present in each ventricle at the end of ventricular diastole. • Preload: load on the muscle in the relaxed state. Indices of left ventricular • Normal amount: 120-130 ml, can be increased during diastole preload: (filling of ventricles) to a volume of (120-130mL). 1- Left ventricle end diastolic • Applying preload to a muscle causes: volume Or pressure (LVEDV) 1- The muscle to stretch. 2- Right atrial pressure 2- The muscle to develop passive tension. More relaxation time, more filling, more volume. EDV Depends on: A- Filling time: the duration of ventricular diastole. B- Venous return: the rate of blood flow during ventricular diastole. 3 Stroke Volume and Factors Affecting it 1- End diastolic volume (EDV) 2- End systolic volume (ESV) (Residual): (Preload): It is: volume of blood present (that remains) in Mechanism: each ventricle at the end of ventricular systole. -

Determination of Cardiac Output by Equating Venous Return Curves with Cardiac Response Curves
Determination of Cardiac Output By Equating Ve- nous Return Curves With Cardiac Response Curves1 ARTHUR C. GUYTQN From the Department of Physiology and Biophysics, School of Medicine, University of Mississippi, University, Mississippi HE CONCEPT that the heart responds with increasing cardiac output when there occurs increasing venous return was popularized by Starling and, in- deed, has come to be known as Starling’s law. There are many different forms in which Starling’s law can be expressed,including the relationship of cardiac output to right atria1 pressure, the relationship of cardiac output to the degree of distention of the right ventricle at the end of diastole, the relationship of cardiac work to right atria1 pressure or right ventricular distention, the relationship of left ventricular work to right atria1 pressure or right ventricular distention, etc. For the determina- tion of cardiac output, the form of Starling’s law which will be used in the present discussion is the relationship of cardiac output to mean right atria1 pressure, and this type of cruve will be called the “cardiac responsecurve” to right atria1 pressure. It is well known that many factors in the peripheral circulatory system combine together to determine the rate of venous return to the heart. These include the quantity of blood available, the degree of vascular resistance in various parts of the peripheral circulatory system, and the back pressure from the right atrium. It is with these factors that this paper is especially concerned, and it is hoped that this presentation will demonstrate how cardiac output is determined by equating the peripheral circulatory factors with the cardiac responsecurves. -
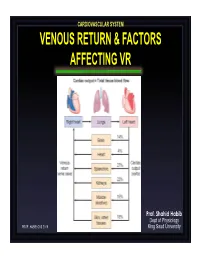
Venous Return & Factors Affecting Vr
CARDIOVASCULAR SYSTEM VENOUS RETURN & FACTORS AFFECTING VR Prof. Shahid Habib Dept of Physiology PROF. HABIB CVS 2019 King Saud University OBJECTIVES At the end of the lecture you should be able to ….. Discuss functions of the veins as blood reservoirs. Describe measurement of central venous pressure (CVP) and state its physiological and clinical significance. State determinants of venous return and explain how they influence venous return. Define mean systemic filling pressure, give its normal value and describe the factors which affect it. Explain the effect of gravity on venous pressure and explain pathophysiology of varicose veins. Describe vascular and cardiac function curves under physiological and pathophysiological conditions. DISTRIBUTION OF BLOOD Capacitance Vessels Veins are blood reservoirs When the body is at rest and many of the capillaries are closed, the capacity of the venous reservoir is increased as extra blood bypasses the capillaries and enters the veins. When this extra volume of blood stretches the veins, the blood moves forward through the veins more slowly because the total cross sectional area of the veins has increased as a result of the stretching. Therefore, blood spends more time in the veins. When the stored blood is needed, such as during exercise, extrinsic factors reduce the capacity of the venous reservoir and drive the extra blood from the veins to the heart so that it can be pumped to the tissues. Rt. Lt. At At Right Left ven ven Large veins Large arteries CAPACITANCE VESSELS EXCHANGE VESSELS RESISTANCE VESSELS Venules Capillaries Arterioles Vascular circuit • all 3 layers are present, but thinner than in •In varicose veins, arteries of corresponding size (external blood pools because diameter). -

Webinar Ch 8
Animal Anatomy and Physiology 1 Webinar Chapter 8 Cardiovascular System The Cardiovascular System Chapter 8 Pages 205-219 Textbook Learning Objectives Chapter 8 – Page 205 • List and describe the layers of the heart wall • List the chambers of the heart and describe the path of blood flow through the heart • Describe the structure and locations of the heart valves • Differentiate between systole and diastole • Describe the process of depolarization and repolarization of cardiac muscle cells • Describe the pathway of the electrical impulse generated by the SA node • List and describe the unique anatomical features of the fetal circulatory system • List the events responsible for the heart sounds heard on auscultation • List the factors that influence heart rate and cardiac output • Describe the relationship between cardiac output, heart rate, and stroke volume • Describe the structures of arteries, capillaries, and veins • List the major arteries and veins that travel from the heart to the systemic circulation • List the names and locations of veins commonly used for venipuncture in animals Cardiovascular System Heart Blood Vessels Blood YourDogsHeart.com http://www.yourdogsheart.com/ Your Cat’s Heart http://www.pethealthnetwork.com/cat-health/your- cat%E2%80%99s-heart-feline-heart-disease Great Human Heart A&P website http://www.nhlbi.nih.gov/health/health-topics/topics/hhw/ Internal Medicine The KEY Is Cellular Health Cellular Health • Healthy Cells = Health Animal Body • Diseased Cells = Diseased Animal Body • Too Many Diseased Cells -

Approach to Shock.” These Podcasts Are Designed to Give Medical Students an Overview of Key Topics in Pediatrics
PedsCases Podcast Scripts This is a text version of a podcast from Pedscases.com on “Approach to Shock.” These podcasts are designed to give medical students an overview of key topics in pediatrics. The audio versions are accessible on iTunes or at www.pedcases.com/podcasts. Approach to Shock Developed by Dr. Dustin Jacobson and Dr Suzanne Beno for PedsCases.com. December 20, 2016 My name is Dustin Jacobson, a 3rd year pediatrics resident from the University of Toronto. This podcast was supervised by Dr. Suzanne Beno, a staff physician in the division of Pediatric Emergency Medicine at the University of Toronto. Today, we’ll discuss an approach to shock in children. First, we’ll define shock and understand it’s pathophysiology. Next, we’ll examine the subclassifications of shock. Last, we’ll review some basic and more advanced treatment for shock But first, let’s start with a case. Jonny is a 6-year-old male who presents with lethargy that is preceded by 2 days of a diarrheal illness. He has not urinated over the previous 24 hours. On assessment, he is tachycardic and hypotensive. He is febrile at 40 degrees Celsius, and is moaning on assessment, but spontaneously breathing. We’ll revisit this case including evaluation and management near the end of this podcast. The term “shock” is essentially a ‘catch-all’ phrase that refers to a state of inadequate oxygen or nutrient delivery for tissue metabolic demand. This broad definition incorporates many causes that eventually lead to this end-stage state. Basic oxygen delivery is determined by cardiac output and content of oxygen in the blood.