Online Supplementary Material
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Aberrant Modulation of Ribosomal Protein S6 Phosphorylation Confers Acquired Resistance to MAPK Pathway Inhibitors in BRAF-Mutant Melanoma
www.nature.com/aps ARTICLE Aberrant modulation of ribosomal protein S6 phosphorylation confers acquired resistance to MAPK pathway inhibitors in BRAF-mutant melanoma Ming-zhao Gao1,2, Hong-bin Wang1,2, Xiang-ling Chen1,2, Wen-ting Cao1,LiFu1, Yun Li1, Hai-tian Quan1,2, Cheng-ying Xie1,2 and Li-guang Lou1,2 BRAF and MEK inhibitors have shown remarkable clinical efficacy in BRAF-mutant melanoma; however, most patients develop resistance, which limits the clinical benefit of these agents. In this study, we found that the human melanoma cell clones, A375-DR and A375-TR, with acquired resistance to BRAF inhibitor dabrafenib and MEK inhibitor trametinib, were cross resistant to other MAPK pathway inhibitors. In these resistant cells, phosphorylation of ribosomal protein S6 (rpS6) but not phosphorylation of ERK or p90 ribosomal S6 kinase (RSK) were unable to be inhibited by MAPK pathway inhibitors. Notably, knockdown of rpS6 in these cells effectively downregulated G1 phase-related proteins, including RB, cyclin D1, and CDK6, induced cell cycle arrest, and inhibited proliferation, suggesting that aberrant modulation of rpS6 phosphorylation contributed to the acquired resistance. Interestingly, RSK inhibitor had little effect on rpS6 phosphorylation and cell proliferation in resistant cells, whereas P70S6K inhibitor showed stronger inhibitory effects on rpS6 phosphorylation and cell proliferation in resistant cells than in parental cells. Thus regulation of rpS6 phosphorylation, which is predominantly mediated by BRAF/MEK/ERK/RSK signaling in parental cells, was switched to mTOR/ P70S6K signaling in resistant cells. Furthermore, mTOR inhibitors alone overcame acquired resistance and rescued the sensitivity of the resistant cells when combined with BRAF/MEK inhibitors. -

Allele-Specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish
Allele-specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish by Ailu Chen A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama August 1, 2015 Keywords: catfish, interspecific hybrids, allele-specific expression, ribosomal protein Copyright 2015 by Ailu Chen Approved by Zhanjiang Liu, Chair, Professor, School of Fisheries, Aquaculture and Aquatic Sciences Nannan Liu, Professor, Entomology and Plant Pathology Eric Peatman, Associate Professor, School of Fisheries, Aquaculture and Aquatic Sciences Aaron M. Rashotte, Associate Professor, Biological Sciences Abstract Interspecific hybridization results in a vast reservoir of allelic variations, which may potentially contribute to phenotypical enhancement in the hybrids. Whether the allelic variations are related to the downstream phenotypic differences of interspecific hybrid is still an open question. The recently developed genome-wide allele-specific approaches that harness high- throughput sequencing technology allow direct quantification of allelic variations and gene expression patterns. In this work, I investigated allele-specific expression (ASE) pattern using RNA-Seq datasets generated from interspecific catfish hybrids. The objective of the study is to determine the ASE genes and pathways in which they are involved. Specifically, my study investigated ASE-SNPs, ASE-genes, parent-of-origins of ASE allele and how ASE would possibly contribute to heterosis. My data showed that ASE was operating in the interspecific catfish system. Of the 66,251 and 177,841 SNPs identified from the datasets of the liver and gill, 5,420 (8.2%) and 13,390 (7.5%) SNPs were identified as significant ASE-SNPs, respectively. -

Mtorc1 Controls Mitochondrial Activity and Biogenesis Through 4E-BP-Dependent Translational Regulation
Cell Metabolism Article mTORC1 Controls Mitochondrial Activity and Biogenesis through 4E-BP-Dependent Translational Regulation Masahiro Morita,1,2 Simon-Pierre Gravel,1,2 Vale´ rie Che´ nard,1,2 Kristina Sikstro¨ m,3 Liang Zheng,4 Tommy Alain,1,2 Valentina Gandin,5,7 Daina Avizonis,2 Meztli Arguello,1,2 Chadi Zakaria,1,2 Shannon McLaughlan,5,7 Yann Nouet,1,2 Arnim Pause,1,2 Michael Pollak,5,6,7 Eyal Gottlieb,4 Ola Larsson,3 Julie St-Pierre,1,2,* Ivan Topisirovic,5,7,* and Nahum Sonenberg1,2,* 1Department of Biochemistry 2Goodman Cancer Research Centre McGill University, Montreal, QC H3A 1A3, Canada 3Department of Oncology-Pathology, Karolinska Institutet, Stockholm, 171 76, Sweden 4Cancer Research UK, The Beatson Institute for Cancer Research, Switchback Road, Glasgow G61 1BD, Scotland, UK 5Lady Davis Institute for Medical Research 6Cancer Prevention Center, Sir Mortimer B. Davis-Jewish General Hospital McGill University, Montreal, QC H3T 1E2, Canada 7Department of Oncology, McGill University, Montreal, QC H2W 1S6, Canada *Correspondence: [email protected] (J.S.-P.), [email protected] (I.T.), [email protected] (N.S.) http://dx.doi.org/10.1016/j.cmet.2013.10.001 SUMMARY ATP under physiological conditions in mammals and play a crit- ical role in overall energy balance (Vander Heiden et al., 2009). mRNA translation is thought to be the most energy- The mechanistic/mammalian target of rapamycin (mTOR) is a consuming process in the cell. Translation and serine/threonine kinase that has been implicated in a variety of energy metabolism are dysregulated in a variety of physiological processes and pathological states (Zoncu et al., diseases including cancer, diabetes, and heart 2011). -

Differential Requirement of the Ribosomal Protein S6 And
Plant Pathology and Microbiology Publications Plant Pathology and Microbiology 5-2017 Differential Requirement of the Ribosomal Protein S6 and Ribosomal Protein S6 Kinase for Plant- Virus Accumulation and Interaction of S6 Kinase with Potyviral VPg Minna-Liisa Rajamäki University of Helsinki Dehui Xi Sichuan University Sidona Sikorskaite-Gudziuniene University of Helsinki Jari P. T. Valkonen University of Helsinki Follow this and additional works at: http://lib.dr.iastate.edu/plantpath_pubs StevePanr tA of. W thehithAgramicultural Science Commons, Agriculture Commons, Plant Breeding and Genetics CIowommona State Usn, iaverndsit they, swPhithlanatm@i Pathoastalote.geduy Commons The ompc lete bibliographic information for this item can be found at http://lib.dr.iastate.edu/ plantpath_pubs/211. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Plant Pathology and Microbiology at Iowa State University Digital Repository. It has been accepted for inclusion in Plant Pathology and Microbiology Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Differential Requirement of the Ribosomal Protein S6 and Ribosomal Protein S6 Kinase for Plant-Virus Accumulation and Interaction of S6 Kinase with Potyviral VPg Abstract Ribosomal protein S6 (RPS6) is an indispensable plant protein regulated, in part, by ribosomal protein S6 kinase (S6K) which, in turn, is a key regulator of plant responses to stresses and developmental cues. Increased expression of RPS6 was detected in Nicotiana benthamiana during infection by diverse plant viruses. Silencing of the RPS6and S6K genes in N. -

Supplementary Table 2
Supplementary Table 2. Differentially Expressed Genes following Sham treatment relative to Untreated Controls Fold Change Accession Name Symbol 3 h 12 h NM_013121 CD28 antigen Cd28 12.82 BG665360 FMS-like tyrosine kinase 1 Flt1 9.63 NM_012701 Adrenergic receptor, beta 1 Adrb1 8.24 0.46 U20796 Nuclear receptor subfamily 1, group D, member 2 Nr1d2 7.22 NM_017116 Calpain 2 Capn2 6.41 BE097282 Guanine nucleotide binding protein, alpha 12 Gna12 6.21 NM_053328 Basic helix-loop-helix domain containing, class B2 Bhlhb2 5.79 NM_053831 Guanylate cyclase 2f Gucy2f 5.71 AW251703 Tumor necrosis factor receptor superfamily, member 12a Tnfrsf12a 5.57 NM_021691 Twist homolog 2 (Drosophila) Twist2 5.42 NM_133550 Fc receptor, IgE, low affinity II, alpha polypeptide Fcer2a 4.93 NM_031120 Signal sequence receptor, gamma Ssr3 4.84 NM_053544 Secreted frizzled-related protein 4 Sfrp4 4.73 NM_053910 Pleckstrin homology, Sec7 and coiled/coil domains 1 Pscd1 4.69 BE113233 Suppressor of cytokine signaling 2 Socs2 4.68 NM_053949 Potassium voltage-gated channel, subfamily H (eag- Kcnh2 4.60 related), member 2 NM_017305 Glutamate cysteine ligase, modifier subunit Gclm 4.59 NM_017309 Protein phospatase 3, regulatory subunit B, alpha Ppp3r1 4.54 isoform,type 1 NM_012765 5-hydroxytryptamine (serotonin) receptor 2C Htr2c 4.46 NM_017218 V-erb-b2 erythroblastic leukemia viral oncogene homolog Erbb3 4.42 3 (avian) AW918369 Zinc finger protein 191 Zfp191 4.38 NM_031034 Guanine nucleotide binding protein, alpha 12 Gna12 4.38 NM_017020 Interleukin 6 receptor Il6r 4.37 AJ002942 -

Inhibition of the MID1 Protein Complex
Matthes et al. Cell Death Discovery (2018) 4:4 DOI 10.1038/s41420-017-0003-8 Cell Death Discovery ARTICLE Open Access Inhibition of the MID1 protein complex: a novel approach targeting APP protein synthesis Frank Matthes1,MoritzM.Hettich1, Judith Schilling1, Diana Flores-Dominguez1, Nelli Blank1, Thomas Wiglenda2, Alexander Buntru2,HannaWolf1, Stephanie Weber1,InaVorberg 1, Alina Dagane2, Gunnar Dittmar2,3,ErichWanker2, Dan Ehninger1 and Sybille Krauss1 Abstract Alzheimer’s disease (AD) is characterized by two neuropathological hallmarks: senile plaques, which are composed of amyloid-β (Aβ) peptides, and neurofibrillary tangles, which are composed of hyperphosphorylated tau protein. Aβ peptides are derived from sequential proteolytic cleavage of the amyloid precursor protein (APP). In this study, we identified a so far unknown mode of regulation of APP protein synthesis involving the MID1 protein complex: MID1 binds to and regulates the translation of APP mRNA. The underlying mode of action of MID1 involves the mTOR pathway. Thus, inhibition of the MID1 complex reduces the APP protein level in cultures of primary neurons. Based on this, we used one compound that we discovered previously to interfere with the MID1 complex, metformin, for in vivo experiments. Indeed, long-term treatment with metformin decreased APP protein expression levels and consequently Aβ in an AD mouse model. Importantly, we have initiated the metformin treatment late in life, at a time-point where mice were in an already progressed state of the disease, and could observe an improved behavioral phenotype. These 1234567890 1234567890 findings together with our previous observation, showing that inhibition of the MID1 complex by metformin also decreases tau phosphorylation, make the MID1 complex a particularly interesting drug target for treating AD. -

The Ribosomal Protein Genes and Minute Loci of Drosophila
Open Access Research2007MarygoldetVolume al. 8, Issue 10, Article R216 The ribosomal protein genes and Minute loci of Drosophila melanogaster Steven J Marygold*, John Roote†, Gunter Reuter‡, Andrew Lambertsson§, Michael Ashburner†, Gillian H Millburn†, Paul M Harrison¶, Zhan Yu¶, Naoya Kenmochi¥, Thomas C Kaufman#, Sally J Leevers* and Kevin R Cook# Addresses: *Growth Regulation Laboratory, Cancer Research UK London Research Institute, Lincoln's Inn Fields, London WC2A 3PX, UK. †Department of Genetics, University of Cambridge, Downing Street, Cambridge CB2 3EH, UK. ‡Institute of Genetics, Biologicum, Martin Luther University Halle-Wittenberg, Weinbergweg, Halle D-06108, Germany. §Institute of Molecular Biosciences, University of Oslo, Blindern, Olso N-0316, Norway. ¶Department of Biology, McGill University, Dr Penfield Ave, Montreal, Quebec H3A 1B1, Canada. ¥Frontier Science Research Center, University of Miyazaki, 5200 Kihara, Kiyotake, Miyazaki 889-1692, Japan. #Department of Biology, Indiana University, E. Third Street, Bloomington, IN 47405-7005, USA. Correspondence: Steven J Marygold. Email: [email protected]. Kevin R Cook. Email: [email protected] Published: 10 October 2007 Received: 17 June 2007 Revised: 10 October 2007 Genome Biology 2007, 8:R216 (doi:10.1186/gb-2007-8-10-r216) Accepted: 10 October 2007 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2007/8/10/R216 © 2007 Marygold et al.; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. -

SCIENCE CHINA the Role of Ribosomal Proteins in the Regulation
SCIENCE CHINA Life Sciences • REVIEW • July 2016 Vol.59 No.7: 656–672 doi: 10.1007/s11427-016-0018-0 The role of ribosomal proteins in the regulation of cell proliferation, tumorigenesis, and genomic integrity Xilong Xu1, Xiufang Xiong1* & Yi Sun1,2,3‡ 1Institute of Translational Medicine, Zhejiang University School of Medicine, Zhejiang 310029, China; 2Collaborative Innovation Center for Diagnosis and Treatment of Infectious Disease, Zhejiang University, Zhejiang 310029, China; 3Division of Radiation and Cancer Biology, Department of Radiation Oncology, University of Michigan, Ann Arbor MI 48109, USA Received March 15, 2016; accepted April 6, 2016; published online June 12, 2016 Ribosomal proteins (RPs), the essential components of the ribosome, are a family of RNA-binding proteins, which play prime roles in ribosome biogenesis and protein translation. Recent studies revealed that RPs have additional extra-ribosomal func- tions, independent of protein biosynthesis, in regulation of diverse cellular processes. Here, we review recent advances in our understanding of how RPs regulate apoptosis, cell cycle arrest, cell proliferation, neoplastic transformation, cell migration and invasion, and tumorigenesis through both MDM2/p53-dependent and p53-independent mechanisms. We also discuss the roles of RPs in the maintenance of genome integrity via modulating DNA damage response and repair. We further discuss mutations or deletions at the somatic or germline levels of some RPs in human cancers as well as in patients of Diamond-Blackfan ane- mia and 5q- syndrome with high susceptibility to cancer development. Moreover, we discuss the potential clinical application, based upon abnormal levels of RPs, in biomarker development for early diagnosis and/or prognosis of certain human cancers. -

Ribosomal Protein S6
MOLECULE PAGE Ribosomal Protein S6 John A. Williams Department of Molecular and Integrative Physiology, University of Michigan e-mail: [email protected] Version 1.0, March 31st, 2021 [DOI: 10.3998/panc.2021.07] Gene Symbol: RPS6 1. General Information and serum stimulated mouse embryo fibroblasts (MEFs) (25). In a variety of specialized cell types Ribosomal protein S6 (rpS6) is one of 33 proteins including muscle, neurons and secretory cells, S6 which along with 18S ribosomal RNA make up the phosphorylation is temporally correlated to the small (40S) subunit of the eukaryotic ribosome. It initiation of protein synthesis. is located in the small head region of the 40S subunit and resides at the interface of the 40S and Considerable success has been achieved in 60S (large) subunits where a groove is thought to understanding the role of different kinases in be the site of new protein synthesis (35). Cross phosphorylating rpS6 (22). The first kinase linking studies suggest rpS6 may interact with identified was in Xenopus oocytes and is now mRNA. rpS6 is an evolutionary conserved protein known as p90 rpS6 Kinase (RSK) (8). This kinase of 236 to 253 amino acids and is the first has an apparent molecular mass of 90 kDa, is discovered and main phosphorylated ribosomal present in mammalian cells and is now known to protein as shown originally by Gressner and Wool be an effector of ERK. Avian and mammalian cells (13). The phosphorylation sites are located in the were then shown to also contain a distinct 70 kDa carboxyl terminus of the protein and have been S6 kinase referred to as p70 S6K but now known mapped in mammals to Ser-235, -236, -240, -244, simply as S6K which can phosphorylate all five and -247 (21). -
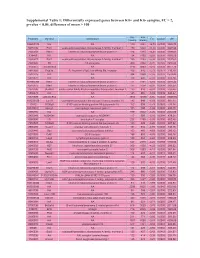
Differentially Expressed Genes Between Kit+ and Kit- Samples, FC
Supplemental Table 1: Differentially expressed genes between Kit+ and Kit- samples, FC > 2, p-value < 0.05, difference of mean > 100 Kit- Kit+ Probeset Symbol Genename FC pvalue diff (mean) (mean) 104280139 NA NA 130 1651 -12,70 0,0000 1520,78 5670239 Ear2 eosinophil-associated, ribonuclease A family, member 2 150 1848 -12,28 0,0000 1697,94 2340358 Ifitm3 interferon induced transmembrane protein 3 126 1315 -10,41 0,0000 1189,02 130465 NA NA 134 1155 -8,60 0,0000 1020,25 2360471 Ear1 eosinophil-associated, ribonuclease A family, member 1 155 1293 -8,34 0,0000 1137,61 7040095 Kit kit oncogene 409 3364 -8,22 0,0145 2955,05 1230347 LOC545854 NA 1216 9893 -8,14 0,0000 8677,76 2810059 Fcgr3a Fc fragment of IgG, low affinity IIIa, receptor 106 816 -7,73 0,0077 710,20 2510725 NA NA 394 2969 -7,53 0,0037 2574,43 5420372 NA NA 128 942 -7,33 0,0000 813,19 101990390 Ifitm2 interferon induced transmembrane protein 2 171 1181 -6,93 0,0000 1010,43 6510075 Ifitm1 interferon induced transmembrane protein 1 152 1054 -6,92 0,0004 901,68 2370286 Slc40a1 solute carrier family 40 (iron-regulated transporter), member 1 133 916 -6,91 0,0000 783,46 5860673 NA NA 143 985 -6,89 0,0239 842,27 6370309 LOC545854 NA 1850 12680 -6,85 0,0000 10829,66 101230129 Ear10 eosinophil-associated, ribonuclease A family, member 10 142 949 -6,68 0,0000 807,11 70112 S100a8 S100 calcium binding protein A8 (calgranulin A) 162 938 -5,79 0,0063 776,16 103780671 Mpeg1 macrophage expressed gene 1 105 598 -5,69 0,0000 493,10 1690184 NA NA 579 3042 -5,26 0,0002 2463,90 2450148 AI324046 expressed -

Accurate Prediction of Kinase-Substrate Networks Using
bioRxiv preprint doi: https://doi.org/10.1101/865055; this version posted December 4, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Accurate Prediction of Kinase-Substrate Networks Using Knowledge Graphs V´ıtNov´aˇcek1∗+, Gavin McGauran3, David Matallanas3, Adri´anVallejo Blanco3,4, Piero Conca2, Emir Mu~noz1,2, Luca Costabello2, Kamalesh Kanakaraj1, Zeeshan Nawaz1, Sameh K. Mohamed1, Pierre-Yves Vandenbussche2, Colm Ryan3, Walter Kolch3,5,6, Dirk Fey3,6∗ 1Data Science Institute, National University of Ireland Galway, Ireland 2Fujitsu Ireland Ltd., Co. Dublin, Ireland 3Systems Biology Ireland, University College Dublin, Belfield, Dublin 4, Ireland 4Department of Oncology, Universidad de Navarra, Pamplona, Spain 5Conway Institute of Biomolecular & Biomedical Research, University College Dublin, Belfield, Dublin 4, Ireland 6School of Medicine, University College Dublin, Belfield, Dublin 4, Ireland ∗ Corresponding authors ([email protected], [email protected]). + Lead author. 1 bioRxiv preprint doi: https://doi.org/10.1101/865055; this version posted December 4, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Phosphorylation of specific substrates by protein kinases is a key control mechanism for vital cell-fate decisions and other cellular pro- cesses. However, discovering specific kinase-substrate relationships is time-consuming and often rather serendipitous. -

Protein S6 Phosphorylation Activation Independently of Ribosomal 1/S6
Mechanistic Target of Rapamycin Complex 1/S6 Kinase 1 Signals Influence T Cell Activation Independently of Ribosomal Protein S6 Phosphorylation This information is current as of September 28, 2021. Robert J. Salmond, Rebecca J. Brownlie, Oded Meyuhas and Rose Zamoyska J Immunol published online 9 October 2015 http://www.jimmunol.org/content/early/2015/10/09/jimmun ol.1501473 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2015/10/09/jimmunol.150147 Material 3.DCSupplemental http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 28, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2015 The Authors All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published October 9, 2015, doi:10.4049/jimmunol.1501473 The Journal of Immunology Mechanistic Target of Rapamycin Complex 1/S6 Kinase 1 Signals Influence T Cell Activation Independently of Ribosomal Protein S6 Phosphorylation Robert J. Salmond,* Rebecca J. Brownlie,* Oded Meyuhas,† and Rose Zamoyska* Ag-dependent activation of naive T cells induces dramatic changes in cellular metabolism that are essential for cell growth, division, and differentiation.