Current Understanding of KATP Channels in Neonatal Diseases: Focus on Insulin Secretion Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sulfonylurea Stimulation of Insulin Secretion Peter Proks,1 Frank Reimann,2 Nick Green,1 Fiona Gribble,2 and Frances Ashcroft1
Sulfonylurea Stimulation of Insulin Secretion Peter Proks,1 Frank Reimann,2 Nick Green,1 Fiona Gribble,2 and Frances Ashcroft1 Sulfonylureas are widely used to treat type 2 diabetes smooth, and skeletal muscle, and some brain neurones. In because they stimulate insulin secretion from pancre- all these tissues, opening of KATP channels in response to atic -cells. They primarily act by binding to the SUR metabolic stress leads to inhibition of electrical activity. subunit of the ATP-sensitive potassium (KATP) channel Thus they are involved in the response to both cardiac and and inducing channel closure. However, the channel is cerebral ischemia (2). They are also important in neuronal still able to open to a limited extent when the drug is regulation of glucose homeostasis (3), seizure protection bound, so that high-affinity sulfonylurea inhibition is not complete, even at saturating drug concentrations. (4), and the control of vascular smooth muscle tone (and, thereby, blood pressure) (5). KATP channels are also found in cardiac, skeletal, and smooth muscle, but in these tissues are composed of The KATP channel is a hetero-octameric complex of two different SUR subunits that confer different drug different types of protein subunits: an inwardly rectifying sensitivities. Thus tolbutamide and gliclazide block Kϩ channel, Kir6.x, and a sulfonylurea receptor, SUR (6,7).  ϩ channels containing SUR1 ( -cell type), but not SUR2 Kir6.x belongs to the family of inwardly rectifying K (cardiac, smooth muscle types), whereas glibenclamide, (Kir) channels and assembles as a tetramer to form the glimepiride, repaglinide, and meglitinide block both types of channels. -

The Genetic Basis of Congenital Hyperinsulinism Chela James, Ritika K Kapoor, Dunia Ismail, Khalid Hussain
The genetic basis of congenital hyperinsulinism Chela James, Ritika K Kapoor, Dunia Ismail, Khalid Hussain To cite this version: Chela James, Ritika K Kapoor, Dunia Ismail, Khalid Hussain. The genetic basis of congeni- tal hyperinsulinism. Journal of Medical Genetics, BMJ Publishing Group, 2009, 46 (5), pp.289. 10.1136/jmg.2008.064337. hal-00552677 HAL Id: hal-00552677 https://hal.archives-ouvertes.fr/hal-00552677 Submitted on 6 Jan 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. The genetic basis of congenital hyperinsulinism Chela James, Ritika R Kapoor, Dunia Ismail, Khalid Hussain London Centre for Paediatric Endocrinology and Metabolism, Great Ormond Street Hospital for Children NHS Trust, London WC1N 3JH, and The Institute of Child Health, University College London WC1N 1EH UK. Correspondence to Dr. K. Hussain Developmental Endocrinology Research Group Clinical and Molecular Genetics Unit Institute of Child Health University College London 30 Guilford Street, London WC1N 1EH. Tele: ++44 (0)20 7 905 2128 Fax: ++44 (0)20 7 404 6191 Email [email protected] The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non-exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Journal of Medical Genetics and any other BMJPGL products to exploit all subsidiary rights, as set out in our licence (http://jmg.bmj.com/iforalicence.pdf). -

Transcriptional and Post-Transcriptional Regulation of ATP-Binding Cassette Transporter Expression
Transcriptional and Post-transcriptional Regulation of ATP-binding Cassette Transporter Expression by Aparna Chhibber DISSERTATION Submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Pharmaceutical Sciences and Pbarmacogenomies in the Copyright 2014 by Aparna Chhibber ii Acknowledgements First and foremost, I would like to thank my advisor, Dr. Deanna Kroetz. More than just a research advisor, Deanna has clearly made it a priority to guide her students to become better scientists, and I am grateful for the countless hours she has spent editing papers, developing presentations, discussing research, and so much more. I would not have made it this far without her support and guidance. My thesis committee has provided valuable advice through the years. Dr. Nadav Ahituv in particular has been a source of support from my first year in the graduate program as my academic advisor, qualifying exam committee chair, and finally thesis committee member. Dr. Kathy Giacomini graciously stepped in as a member of my thesis committee in my 3rd year, and Dr. Steven Brenner provided valuable input as thesis committee member in my 2nd year. My labmates over the past five years have been incredible colleagues and friends. Dr. Svetlana Markova first welcomed me into the lab and taught me numerous laboratory techniques, and has always been willing to act as a sounding board. Michael Martin has been my partner-in-crime in the lab from the beginning, and has made my days in lab fly by. Dr. Yingmei Lui has made the lab run smoothly, and has always been willing to jump in to help me at a moment’s notice. -

Whole-Exome Sequencing Identifies Novel Mutations in ABC Transporter
Liu et al. BMC Pregnancy and Childbirth (2021) 21:110 https://doi.org/10.1186/s12884-021-03595-x RESEARCH ARTICLE Open Access Whole-exome sequencing identifies novel mutations in ABC transporter genes associated with intrahepatic cholestasis of pregnancy disease: a case-control study Xianxian Liu1,2†, Hua Lai1,3†, Siming Xin1,3, Zengming Li1, Xiaoming Zeng1,3, Liju Nie1,3, Zhengyi Liang1,3, Meiling Wu1,3, Jiusheng Zheng1,3* and Yang Zou1,2* Abstract Background: Intrahepatic cholestasis of pregnancy (ICP) can cause premature delivery and stillbirth. Previous studies have reported that mutations in ABC transporter genes strongly influence the transport of bile salts. However, to date, their effects are still largely elusive. Methods: A whole-exome sequencing (WES) approach was used to detect novel variants. Rare novel exonic variants (minor allele frequencies: MAF < 1%) were analyzed. Three web-available tools, namely, SIFT, Mutation Taster and FATHMM, were used to predict protein damage. Protein structure modeling and comparisons between reference and modified protein structures were performed by SWISS-MODEL and Chimera 1.14rc, respectively. Results: We detected a total of 2953 mutations in 44 ABC family transporter genes. When the MAF of loci was controlled in all databases at less than 0.01, 320 mutations were reserved for further analysis. Among these mutations, 42 were novel. We classified these loci into four groups (the damaging, probably damaging, possibly damaging, and neutral groups) according to the prediction results, of which 7 novel possible pathogenic mutations were identified that were located in known functional genes, including ABCB4 (Trp708Ter, Gly527Glu and Lys386Glu), ABCB11 (Gln1194Ter, Gln605Pro and Leu589Met) and ABCC2 (Ser1342Tyr), in the damaging group. -

Whole Exome Sequencing Analysis of ABCC8 and ABCD2 Genes Associating with Clinical Course of Breast Carcinoma
Physiol. Res. 64 (Suppl. 4): S549-S557, 2015 Whole Exome Sequencing Analysis of ABCC8 and ABCD2 Genes Associating With Clinical Course of Breast Carcinoma P. SOUCEK1,2, V. HLAVAC1,2,3, K. ELSNEROVA1,2,3, R. VACLAVIKOVA2, R. KOZEVNIKOVOVA4, K. RAUS5 1Biomedical Center, Faculty of Medicine in Pilsen, Charles University in Prague, Pilsen, Czech Republic, 2Toxicogenomics Unit, National Institute of Public Health, Prague, Czech Republic, 3Third Faculty of Medicine, Charles University in Prague, Prague, Czech Republic, 4Department of Oncosurgery, Medicon, Prague, Czech Republic, 5Institute for the Care for Mother and Child, Prague, Czech Republic Received September 17, 2015 Accepted October 2, 2015 Summary Charles University in Prague, Alej Svobody 76, 323 00 Pilsen, The aim of the present study was to introduce methods for Czech Republic. E-mail: [email protected] exome sequencing of two ATP-binding cassette (ABC) transporters ABCC8 and ABCD2 recently suggested to play Introduction a putative role in breast cancer progression and prognosis of patients. We performed next generation sequencing targeted at Breast cancer is the most common cancer in analysis of all exons in ABCC8 and ABCD2 genes and surrounding women and caused 471,000 deaths worldwide in 2013 noncoding sequences in blood DNA samples from 24 patients (Global Burden of Disease Cancer Collaboration 2015). with breast cancer. The revealed alterations were characterized A number of cellular processes that in some cases lead to the by in silico tools. We then compared the most frequent tumor resistance limits efficacy of breast cancer therapy. functionally relevant polymorphism rs757110 in ABCC8 with Multidrug resistance (MDR) to a variety of chemotherapy clinical data of patients. -
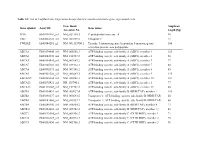
List of Taqman Gene Expression Assays That Were Used to Determine Gene Expression Levels
Table S1: List of TaqMan Gene Expression Assays that were used to determine gene expression levels Gene Bank Amplicon Gene symbol Assay ID Gene name Accession No. length [bp] PPIA Hs99999904_m1 NM_021130.3 Peptidylprolyl isomerase A 98 UBC Hs00824723_m1 NM_021009.5 Ubiquitin C 71 YWHAZ Hs03044281_g1 NM_001135700.1 Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase 106 activation protein, zeta polypeptide ABCA1 Hs00194045_m1 NM_005502.3 ATP-binding cassette, sub-family A (ABC1), member 1 125 ABCA2 Hs00242232_m1 NM_212533.2 ATP-binding cassette, sub-family A (ABC1), member 2 58 ABCA3 Hs00184543_m1 NM_001089.2 ATP-binding cassette, sub-family A (ABC1), member 3 77 ABCA7 Hs00185303_m1 NM_019112.3 ATP-binding cassette, sub-family A (ABC1), member 7 80 ABCA8 Hs00992371_m1 NM_007168.2 ATP-binding cassette, sub-family A (ABC1), member 8 85 ABCA9 Hs00329320_m1 NM_080283.3 ATP-binding cassette, sub-family A (ABC1), member 9 145 ABCA10a Hs00365268_m1 NM_080282.3 ATP-binding cassette, sub-family A (ABC1), member 10 127 ABCA12 Hs00292421_m1 NR_103740.1 ATP-binding cassette, sub-family A (ABC1), member 1 77 ABCA13 Hs01110169_m1 NM_152701.3 ATP-binding cassette, sub-family A (ABC1), member 13 80 ABCB1 Hs00184491_m1 NM_000927.4 ATP-binding cassette, sub-family B (MDR/TAP), member 1 110 ABCB2 Hs00388677_m1 NM_000593.5 Transporter 1, ATP-binding cassette, sub-family B (MDR/TAP) 60 ABCB3 Hs00241060_m1 NM_018833.2 Transporter 2, ATP-binding cassette, sub-family B (MDR/TAP) 66 ABCB4 Hs00240956_m1 NM_018850.2 ATP-binding cassette, sub-family B (MDR/TAP), member -

The Insulin Secretory Granule Is the Major Site of KATP Channels of the Endocrine Pancreas Xuehui Geng,1 Lehong Li,1 Simon Watkins,1 Paul D
The Insulin Secretory Granule Is the Major Site of KATP Channels of the Endocrine Pancreas Xuehui Geng,1 Lehong Li,1 Simon Watkins,1 Paul D. Robbins,2 and Peter Drain1 an on/off switch. When on, potassium ions flow out With ATP sites on Kir6.2 that inhibit activity and ADP sites on SUR1 that antagonize the inhibition, ATP- through the channel electrically hyperpolarizing the -cell, sensitive potassium channels (KATP channels) are de- putting a brake on the signal flow controlling insulin signed as exquisite sensors of adenine nucleotide levels secretion (11). When off, the KATP channel, which is that signal changes in glucose metabolism. If pancreatic inhibited by sulfonylureas or by the increased ATP/ADP KATP channels localize to the insulin secretory granule, ratio from glucose metabolism (12,13), removes this they would be well positioned to transduce changes in brake, allowing initiation of insulin release by elevating glucose metabolism into changes in granule transport intracellular calcium (14–17), which triggers the exocy- and exocytosis. Tests for pancreatic K channels lo- ATP totic fusion of insulin granule and -cell membrane. calized to insulin secretory granules led to the following observations: fluorescent sulfonylureas that bind the The calcium signal, however, is insufficient for regulat- ing insulin release in response to glucose under typical pancreatic KATP channel specifically label intracellular punctate structures in cells of the endocrine pancreas. conditions (11,18). While pharmacologically either open- The fluorescent glibenclamides colocalize with Ins-C- ing or inhibiting the plasma membrane KATP channel, GFP, a live-cell fluorescent reporter of insulin granules. maneuvers that elevate not only intracellular calcium but Expression of either SUR1-GFP or Kir6.2-GFP fusion also glucose metabolism are necessary to confer glucose proteins, but not expression of GFP alone, directs GFP dose dependency to stimulated insulin release. -

Cardiovascular Diseases Genetic Testing Program Information
Cardiovascular Diseases Genetic Testing Program Description: Congenital Heart Disease Panels We offer comprehensive gene panels designed to • Congenital Heart Disease Panel (187 genes) diagnose the most common genetic causes of hereditary • Heterotaxy Panel (114 genes) cardiovascular diseases. Testing is available for congenital • RASopathy/Noonan Spectrum Disorders Panel heart malformation, cardiomyopathy, arrythmia, thoracic (31 genes) aortic aneurysm, pulmonary arterial hypertension, Marfan Other Panels syndrome, and RASopathy/Noonan spectrum disorders. • Pulmonary Arterial Hypertension (PAH) Panel Hereditary cardiovascular disease is caused by variants in (20 genes) many different genes, and may be inherited in an autosomal dominant, autosomal recessive, or X-linked manner. Other Indications: than condition-specific panels, we also offer single gene Panels: sequencing for any gene on the panels, targeted variant • Confirmation of genetic diagnosis in a patient with analysis, and targeted deletion/duplication analysis. a clinical diagnosis of cardiovascular disease Tests Offered: • Carrier or pre-symptomatic diagnosis identification Arrythmia Panels in individuals with a family history of cardiovascular • Comprehensive Arrhythmia Panel (81 genes) disease of unknown genetic basis • Atrial Fibrillation (A Fib) Panel (28 genes) Gene Specific Sequencing: • Atrioventricular Block (AV Block) Panel (7 genes) • Confirmation of genetic diagnosis in a patient with • Brugada Syndrome Panel (21 genes) cardiovascular disease and in whom a specific -

ABCC8 Mrna Expression Is an Independent Prognostic Factor For
www.nature.com/scientificreports OPEN ABCC8 mRNA expression is an independent prognostic factor for glioma and can predict chemosensitivity Kaijia Zhou1, Yanwei Liu1, Zheng Zhao1, Yinyuan Wang1, Lijie Huang1, Ruichao Chai1, Guanzhang Li1 & Tao Jiang1,2,3,4* Glioma is the most common primary intracranial tumor and is associated with very low survival rates. The development of reliable biomarkers can help to elucidate the molecular mechanisms involved in glioma development. Here the expression of ABCC8 mRNA, clinical characteristics, and survival information based on 1893 glioma samples from four independent databases were analyzed. The expression patterns of ABCC8 mRNA were compared by a Chi square test. The overall survival rate of gliomas was evaluated according to the expression level of ABCC8 mRNA. The prognostic value of this marker in gliomas was tested using Cox single factor and multi factor regression analyses. We found patients with low WHO grade, oligodendrocytoma, low molecular grade, IDH mutation, and 1p19q combined deletion had high ABCC8 mRNA expression. The patients with high expression of ABCC8 mRNA had longer survival. ABCC8 mRNA expression was a new independent prognostic index for glioma. Temozolomide chemotherapy was an independent index to prolong overall survival in high ABCC8 mRNA expression glioma patients, whereas in patients with low expression, there was no signifcant diference. So ABCC8 mRNA expression could be an independent prognostic indicator for glioma patients and could predict the sensitivity of glioma to temozolomide. Glioma is the most common primary intracranial tumor, accounting for 81% of malignant brain tumors1. World Health Organization (WHO) grade 4 glioblastoma is the most malignant form of glioma with a 5-year relative survival rate of 5%2. -

Whole Exome Sequencing Analysis of ABCC8 and ABCD2 Genes Associating with Clinical Course of Breast Carcinoma
Physiol. Res. 64 (Suppl. 4): S549-S557, 2015 https://doi.org/10.33549/physiolres.933212 Whole Exome Sequencing Analysis of ABCC8 and ABCD2 Genes Associating With Clinical Course of Breast Carcinoma P. SOUCEK1,2, V. HLAVAC1,2,3, K. ELSNEROVA1,2,3, R. VACLAVIKOVA2, R. KOZEVNIKOVOVA4, K. RAUS5 1Biomedical Center, Faculty of Medicine in Pilsen, Charles University in Prague, Pilsen, Czech Republic, 2Toxicogenomics Unit, National Institute of Public Health, Prague, Czech Republic, 3Third Faculty of Medicine, Charles University in Prague, Prague, Czech Republic, 4Department of Oncosurgery, Medicon, Prague, Czech Republic, 5Institute for the Care for Mother and Child, Prague, Czech Republic Received September 17, 2015 Accepted October 2, 2015 Summary Charles University in Prague, Alej Svobody 76, 323 00 Pilsen, The aim of the present study was to introduce methods for Czech Republic. E-mail: [email protected] exome sequencing of two ATP-binding cassette (ABC) transporters ABCC8 and ABCD2 recently suggested to play Introduction a putative role in breast cancer progression and prognosis of patients. We performed next generation sequencing targeted at Breast cancer is the most common cancer in analysis of all exons in ABCC8 and ABCD2 genes and surrounding women and caused 471,000 deaths worldwide in 2013 noncoding sequences in blood DNA samples from 24 patients (Global Burden of Disease Cancer Collaboration 2015). with breast cancer. The revealed alterations were characterized A number of cellular processes that in some cases lead to the by in silico tools. We then compared the most frequent tumor resistance limits efficacy of breast cancer therapy. functionally relevant polymorphism rs757110 in ABCC8 with Multidrug resistance (MDR) to a variety of chemotherapy clinical data of patients. -

Human -Endosulfine, a Possible Regulator of Sulfonylurea- Sensitive KATP Channel: Molecular Cloning, Expression and Biological P
Proc. Natl. Acad. Sci. USA Vol. 95, pp. 8387–8391, July 1998 Physiology Human a-endosulfine, a possible regulator of sulfonylurea- sensitive KATP channel: Molecular cloning, expression and biological properties LISA HERON*, ANNE VIRSOLVY*, KARINE PEYROLLIER*, FIONA M. GRIBBLE†,ALPHONSE LE CAM*, FRANCES M. ASHCROFT†, AND DOMINIQUE BATAILLE*‡ *Institut National de la Sante´et de la Recherche Me´dicaleU 376, CHU Arnaud-de-Villeneuve, 371 Avenue du Doyen Gaston Giraud, 34295 Montpellier Cedex 05, France; and †University Laboratory of Physiology, Parks Road, Oxford OX1 3PT, United Kingdom Communicated by Viktor Mutt, Karolinska Institutet, Stockholm, Sweden, April 28, 1998 (received for review February 9, 1998) ABSTRACT Sulfonylureas are a class of drugs commonly the presence of an endogenous regulator of KATP channel used in the management of non-insulin-dependent diabetes activity that interacts with the same site as these drugs. This led mellitus. Their therapeutic action results primarily from their to the purification of a- and b-endosulfine from ovine brain ability to inhibit ATP-sensitive potassium (KATP) channels in (11), two peptides that inhibit binding of sulfonylureas to their the plasma membrane of pancreatic b cells and thereby receptor in vitro (12). During the search for endosulfine (11), stimulate insulin release. A key question is whether an en- a peptide corresponding to the high molecular mass a-form dogenous ligand for the KATP channel exists that is able to was isolated from porcine brain and partially sequenced (13). mimic the inhibitory effects of sulfonylureas. We describe here A partial cDNA sequence of a-endosulfine was determined the cloning of the cDNA encoding human a-endosulfine, a (14) and found to show strong homology with that of a bovine 13-kDa peptide that is a putative candidate for such a role. -

The ABCA Subclass of Mammalian Transporters
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1461 (1999) 395^404 www.elsevier.com/locate/bba Review The ABCA subclass of mammalian transporters Cyril Broccardo, Marie-Francoise Luciani, Giovanna Chimini * Centre d'Immunologie de Marseille-Luminy, Parc Scienti¢que de Luminy, 13288 Marseille Cedex 09, France Received 7 September 1999; accepted 15 September 1999 Abstract We describe here a subclass of mammalian ABC transporters, the ABCA subfamily. This is a unique group that, in contrast to any other human ABC transporters, lacks a structural counterpart in yeast. The structural hallmark of the ABCA subfamily is the presence of a stretch of hydrophobic amino acids thought to span the membrane within the putative regulatory (R) domain. As for today, four ABCA transporters have been fully characterised but 11 ABCA-encoding genes have been identified. ABCA-specific motifs in the nucleotide binding folds can be detected when analysing the conserved sequences among the different members. These motifs may reveal functional constraints exclusive to this group of ABC transporters. ß 1999 Elsevier Science B.V. All rights reserved. Keywords: ABCA subclass; ABC1; ABCR; Consensus motif; Gene family Contents 1. Introduction .......................................................... 396 2. Features of the ABCA class of ABC transporters . ............................ 396 3. The ABCA gene family . ................................................