Infinity Biologix Taqpath SARS-Cov-2 Assay
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Infinity Theorem Is Presented Stating That There Is at Least One Multivalued Series That Diverge to Infinity and Converge to Infinite Finite Values
Open Journal of Mathematics and Physics | Volume 2, Article 75, 2020 | ISSN: 2674-5747 https://doi.org/10.31219/osf.io/9zm6b | published: 4 Feb 2020 | https://ojmp.wordpress.com CX [microresearch] Diamond Open Access The infinity theorem Open Mathematics Collaboration∗† March 19, 2020 Abstract The infinity theorem is presented stating that there is at least one multivalued series that diverge to infinity and converge to infinite finite values. keywords: multivalued series, infinity theorem, infinite Introduction 1. 1, 2, 3, ..., ∞ 2. N =x{ N 1, 2,∞3,}... ∞ > ∈ = { } The infinity theorem 3. Theorem: There exists at least one divergent series that diverge to infinity and converge to infinite finite values. ∗All authors with their affiliations appear at the end of this paper. †Corresponding author: [email protected] | Join the Open Mathematics Collaboration 1 Proof 1 4. S 1 1 1 1 1 1 ... 2 [1] = − + − + − + = (a) 5. Sn 1 1 1 ... 1 has n terms. 6. S = lim+n + S+n + 7. A+ft=er app→ly∞ing the limit in (6), we have S 1 1 1 ... + 8. From (4) and (7), S S 2 = 0 + 2 + 0 + 2 ... 1 + 9. S 2 2 1 1 1 .+.. = + + + + + + 1 10. Fro+m (=7) (and+ (9+), S+ )2 2S . + +1 11. Using (6) in (10), limn+ =Sn 2 2 limn Sn. 1 →∞ →∞ 12. limn Sn 2 + = →∞ 1 13. From (6) a=nd (12), S 2. + 1 14. From (7) and (13), S = 1 1 1 ... 2. + = + + + = (b) 15. S 1 1 1 1 1 ... + 1 16. S =0 +1 +1 +1 +1 +1 1 .. -

The Modal Logic of Potential Infinity, with an Application to Free Choice
The Modal Logic of Potential Infinity, With an Application to Free Choice Sequences Dissertation Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Ethan Brauer, B.A. ∼6 6 Graduate Program in Philosophy The Ohio State University 2020 Dissertation Committee: Professor Stewart Shapiro, Co-adviser Professor Neil Tennant, Co-adviser Professor Chris Miller Professor Chris Pincock c Ethan Brauer, 2020 Abstract This dissertation is a study of potential infinity in mathematics and its contrast with actual infinity. Roughly, an actual infinity is a completed infinite totality. By contrast, a collection is potentially infinite when it is possible to expand it beyond any finite limit, despite not being a completed, actual infinite totality. The concept of potential infinity thus involves a notion of possibility. On this basis, recent progress has been made in giving an account of potential infinity using the resources of modal logic. Part I of this dissertation studies what the right modal logic is for reasoning about potential infinity. I begin Part I by rehearsing an argument|which is due to Linnebo and which I partially endorse|that the right modal logic is S4.2. Under this assumption, Linnebo has shown that a natural translation of non-modal first-order logic into modal first- order logic is sound and faithful. I argue that for the philosophical purposes at stake, the modal logic in question should be free and extend Linnebo's result to this setting. I then identify a limitation to the argument for S4.2 being the right modal logic for potential infinity. -

Cantor on Infinity in Nature, Number, and the Divine Mind
Cantor on Infinity in Nature, Number, and the Divine Mind Anne Newstead Abstract. The mathematician Georg Cantor strongly believed in the existence of actually infinite numbers and sets. Cantor’s “actualism” went against the Aristote- lian tradition in metaphysics and mathematics. Under the pressures to defend his theory, his metaphysics changed from Spinozistic monism to Leibnizian volunta- rist dualism. The factor motivating this change was two-fold: the desire to avoid antinomies associated with the notion of a universal collection and the desire to avoid the heresy of necessitarian pantheism. We document the changes in Can- tor’s thought with reference to his main philosophical-mathematical treatise, the Grundlagen (1883) as well as with reference to his article, “Über die verschiedenen Standpunkte in bezug auf das aktuelle Unendliche” (“Concerning Various Perspec- tives on the Actual Infinite”) (1885). I. he Philosophical Reception of Cantor’s Ideas. Georg Cantor’s dis- covery of transfinite numbers was revolutionary. Bertrand Russell Tdescribed it thus: The mathematical theory of infinity may almost be said to begin with Cantor. The infinitesimal Calculus, though it cannot wholly dispense with infinity, has as few dealings with it as possible, and contrives to hide it away before facing the world Cantor has abandoned this cowardly policy, and has brought the skeleton out of its cupboard. He has been emboldened on this course by denying that it is a skeleton. Indeed, like many other skeletons, it was wholly dependent on its cupboard, and vanished in the light of day.1 1Bertrand Russell, The Principles of Mathematics (London: Routledge, 1992 [1903]), 304. -

Measuring Fractals by Infinite and Infinitesimal Numbers
MEASURING FRACTALS BY INFINITE AND INFINITESIMAL NUMBERS Yaroslav D. Sergeyev DEIS, University of Calabria, Via P. Bucci, Cubo 42-C, 87036 Rende (CS), Italy, N.I. Lobachevsky State University, Nizhni Novgorod, Russia, and Institute of High Performance Computing and Networking of the National Research Council of Italy http://wwwinfo.deis.unical.it/∼yaro e-mail: [email protected] Abstract. Traditional mathematical tools used for analysis of fractals allow one to distinguish results of self-similarity processes after a finite number of iterations. For example, the result of procedure of construction of Cantor’s set after two steps is different from that obtained after three steps. However, we are not able to make such a distinction at infinity. It is shown in this paper that infinite and infinitesimal numbers proposed recently allow one to measure results of fractal processes at different iterations at infinity too. First, the new technique is used to measure at infinity sets being results of Cantor’s proce- dure. Second, it is applied to calculate the lengths of polygonal geometric spirals at different points of infinity. 1. INTRODUCTION During last decades fractals have been intensively studied and applied in various fields (see, for instance, [4, 11, 5, 7, 12, 20]). However, their mathematical analysis (except, of course, a very well developed theory of fractal dimensions) very often continues to have mainly a qualitative character and there are no many tools for a quantitative analysis of their behavior after execution of infinitely many steps of a self-similarity process of construction. Usually, we can measure fractals in a way and can give certain numerical answers to questions regarding fractals only if a finite number of steps in the procedure of their construction has been executed. -
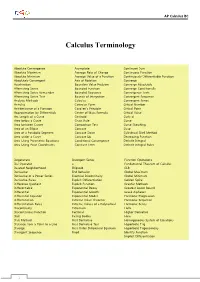
Calculus Terminology
AP Calculus BC Calculus Terminology Absolute Convergence Asymptote Continued Sum Absolute Maximum Average Rate of Change Continuous Function Absolute Minimum Average Value of a Function Continuously Differentiable Function Absolutely Convergent Axis of Rotation Converge Acceleration Boundary Value Problem Converge Absolutely Alternating Series Bounded Function Converge Conditionally Alternating Series Remainder Bounded Sequence Convergence Tests Alternating Series Test Bounds of Integration Convergent Sequence Analytic Methods Calculus Convergent Series Annulus Cartesian Form Critical Number Antiderivative of a Function Cavalieri’s Principle Critical Point Approximation by Differentials Center of Mass Formula Critical Value Arc Length of a Curve Centroid Curly d Area below a Curve Chain Rule Curve Area between Curves Comparison Test Curve Sketching Area of an Ellipse Concave Cusp Area of a Parabolic Segment Concave Down Cylindrical Shell Method Area under a Curve Concave Up Decreasing Function Area Using Parametric Equations Conditional Convergence Definite Integral Area Using Polar Coordinates Constant Term Definite Integral Rules Degenerate Divergent Series Function Operations Del Operator e Fundamental Theorem of Calculus Deleted Neighborhood Ellipsoid GLB Derivative End Behavior Global Maximum Derivative of a Power Series Essential Discontinuity Global Minimum Derivative Rules Explicit Differentiation Golden Spiral Difference Quotient Explicit Function Graphic Methods Differentiable Exponential Decay Greatest Lower Bound Differential -

Attributes of Infinity
International Journal of Applied Physics and Mathematics Attributes of Infinity Kiamran Radjabli* Utilicast, La Jolla, California, USA. * Corresponding author. Email: [email protected] Manuscript submitted May 15, 2016; accepted October 14, 2016. doi: 10.17706/ijapm.2017.7.1.42-48 Abstract: The concept of infinity is analyzed with an objective to establish different infinity levels. It is proposed to distinguish layers of infinity using the diverging functions and series, which transform finite numbers to infinite domain. Hyper-operations of iterated exponentiation establish major orders of infinity. It is proposed to characterize the infinity by three attributes: order, class, and analytic value. In the first order of infinity, the infinity class is assessed based on the “analytic convergence” of the Riemann zeta function. Arithmetic operations in infinity are introduced and the results of the operations are associated with the infinity attributes. Key words: Infinity, class, order, surreal numbers, divergence, zeta function, hyperpower function, tetration, pentation. 1. Introduction Traditionally, the abstract concept of infinity has been used to generically designate any extremely large result that cannot be measured or determined. However, modern mathematics attempts to introduce new concepts to address the properties of infinite numbers and operations with infinities. The system of hyperreal numbers [1], [2] is one of the approaches to define infinite and infinitesimal quantities. The hyperreals (a.k.a. nonstandard reals) *R, are an extension of the real numbers R that contains numbers greater than anything of the form 1 + 1 + … + 1, which is infinite number, and its reciprocal is infinitesimal. Also, the set theory expands the concept of infinity with introduction of various orders of infinity using ordinal numbers. -

Georg Cantor English Version
GEORG CANTOR (March 3, 1845 – January 6, 1918) by HEINZ KLAUS STRICK, Germany There is hardly another mathematician whose reputation among his contemporary colleagues reflected such a wide disparity of opinion: for some, GEORG FERDINAND LUDWIG PHILIPP CANTOR was a corruptor of youth (KRONECKER), while for others, he was an exceptionally gifted mathematical researcher (DAVID HILBERT 1925: Let no one be allowed to drive us from the paradise that CANTOR created for us.) GEORG CANTOR’s father was a successful merchant and stockbroker in St. Petersburg, where he lived with his family, which included six children, in the large German colony until he was forced by ill health to move to the milder climate of Germany. In Russia, GEORG was instructed by private tutors. He then attended secondary schools in Wiesbaden and Darmstadt. After he had completed his schooling with excellent grades, particularly in mathematics, his father acceded to his son’s request to pursue mathematical studies in Zurich. GEORG CANTOR could equally well have chosen a career as a violinist, in which case he would have continued the tradition of his two grandmothers, both of whom were active as respected professional musicians in St. Petersburg. When in 1863 his father died, CANTOR transferred to Berlin, where he attended lectures by KARL WEIERSTRASS, ERNST EDUARD KUMMER, and LEOPOLD KRONECKER. On completing his doctorate in 1867 with a dissertation on a topic in number theory, CANTOR did not obtain a permanent academic position. He taught for a while at a girls’ school and at an institution for training teachers, all the while working on his habilitation thesis, which led to a teaching position at the university in Halle. -

A Mathematical Model of Divine Infinity
A Mathematical Model of Divine Infinity Prof. Eric Steinhart, Department of Philosophy, William Paterson University, Wayne NJ 07470. Email: <[email protected]>, <[email protected]>. Published in 2009 in Theology and Science 7 (3), 261 – 274. ABSTRACT: Mathematics is obviously important in the sciences. And so it is likely to be equally important in any effort that aims to understand God in a scientifically significant way or that aims to clarify the relations between science and theology. The degree to which God has any perfection is absolutely infinite. We use contemporary mathematics to precisely define that absolute infinity. For any perfection, we use transfinite recursion to define an endlessly ascending series of degrees of that perfection. That series rises to an absolutely infinite degree of that perfection. God has that absolutely infinite degree. We focus on the perfections of knowledge, power, and benevolence. Our model of divine infinity thus builds a bridge between mathematics and theology. KEYWORDS: God; mathematics; perfection; infinity; Cantor; transfinite recursion. 1. Introduction All will agree that science makes extensive use of mathematics. At least in physics, scientific progress seems to go hand in hand with increasing formalization. More sophisticated physical theories are also more highly mathematical. And the correlation of progress with increasing formalization also appears sciences like chemistry and biology as well. Mathematics is obviously important in the sciences. And so it is likely to be equally important in any effort that aims to understand God in a scientifically significant way or that aims to clarify the relations between science and theology. Nevertheless, mathematics has seen little application in theology. -

PM752C: Approaching Infinity
INFINITY Prepared for the course team by John Mason Centre for Mathematics Education Project MATHEMATICS UPDATE Course Team Gaynor Arrowsmith, Project Officer, Open University Lynne Burrell, Academic Editor, Open University Leone Burton, External Assessor, Thames Polytechnic Joy Davis, Liaison Adviser, Open University Peter Gates, Author, Open University Pete Griffin, Author, Open University Nick James, Liaison Adviser, Open University Barbara Jaworski, Author, Open University John Mason, Author and Project Leader, Open University Ruth Woolf, Project Secretary, Open University Acknowledgments Project MATHEMATICS UPDATE was funded by a grant from the Department of Education and Science. We are most grateful for comments from Ruth Eagle, Arthur Hanley, Michelle Selinger, Eileen Billington, Gillian Hatch and many others who may not have realised at the time that they were working on parts of this pack. The Open University, Walton Hall, Milton Keynes MK7 6AA. First published 1988. Copyright O 1988 The Open University. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted, in any form or by any means, without written permission from the publisher. Printed in Great Britain by The Open University. Further information on this and other Open University courses may be obtained from the Learning Materials Service Office, Centre for Continuing Education, The Open University, PO Box 188, Milton Keynes MK7 6DH. ISBN 0 335 15918 4 CONTENTS INTRODUCTION AIMS WAYS OF WORKING ENCOUNTERS WITH THE -

Insight Into Pupils' Understanding of Infinity In
INSIGHT INTO PUPILS’ UNDERSTANDING OF INFINITY IN A GEOMETRICAL CONTEXT Darina Jirotková Graham Littler Charles University, Prague, CZ University of Derby, UK Abstract. School students’ intuitive concepts of infinity are gained from personal experiences and in many cases the tacit models built up by them are inconsistent. The paper describes and analyses a series of tasks which were developed to enable the researchers to look into the mental processes used by students when they are thinking about infinity and to help the students to clarify their thoughts on the topic. INTRODUCTION Students’ understanding of infinity is determined by life experiences, on the base of which they have developed tacit models of infinity in their minds. These tacit models (Fischbein, 2001) deal with repeated, everlasting processes, such as going on forever, continued sub-division etc. which are considered mathematically as potential infinity, or by the consideration of a sequence Boero et al (2003) called sequential infinity. Basic concepts of mathematical analysis are usually introduced through limiting processes, which also reinforces the idea of infinity as a process. However, the symbol � is used as a number when tasks about limits and some other concepts of mathematical analysis are solved. When we consider the historical development of infinity, philosophers and mathematicians were dealing with this concept only in its potential form for over 1500 years. Moreno and Waldegg (1991) noted how a grammatical analysis of the use of infinity could be applied to Greek culture. The Greeks used the word ‘infinity’ in a mathematical context only as an adverb, indicating an infinitely continuing action, which is an expression of potential infinity and it was not until 1877that actual infinity, expressed as a noun in mathematics, was defined by Cantor. -

The Strange Properties of the Infinite Power Tower Arxiv:1908.05559V1
The strange properties of the infinite power tower An \investigative math" approach for young students Luca Moroni∗ (August 2019) Nevertheless, the fact is that there is nothing as dreamy and poetic, nothing as radical, subversive, and psychedelic, as mathematics. Paul Lockhart { \A Mathematician's Lament" Abstract In this article we investigate some "unexpected" properties of the \Infinite Power Tower 1" function (or \Tetration with infinite height"): . .. xx y = f(x) = xx where the \tower" of exponentiations has an infinite height. Apart from following an initial personal curiosity, the material collected here is also intended as a potential guide for teachers of high-school/undergraduate students interested in planning an activity of \investigative mathematics in the classroom", where the knowledge is gained through the active, creative and cooperative use of diversified mathematical tools (and some ingenuity). The activity should possibly be carried on with a laboratorial style, with no preclusions on the paths chosen and undertaken by the students and with little or no information imparted from the teacher's desk. The teacher should then act just as a guide and a facilitator. The infinite power tower proves to be particularly well suited to this kind of learning activity, as the student will have to face a challenging function defined through a rather uncommon infinite recursive process. They'll then have to find the right strategies to get around the trickiness of this function and achieve some concrete results, without the help of pre-defined procedures. The mathematical requisites to follow this path are: functions, properties of exponentials and logarithms, sequences, limits and derivatives. -

Pexip Infinity User Guide Welcome to Your Virtual Meeting Room
Pexip Infinity User Guide Welcome to your Virtual Meeting Room Your Virtual Meeting Room (VMR) is a personal space that is always available for you to meet with others over video or audio, and share presentations. You can access your VMR from the Infinity Connect desktop client, a web browser, or any other type of video endpoint. This guide explains all you need to know about: l Using your Virtual Meeting Room to host a conference. l Using Infinity Connect to call into a Virtual Meeting Room or Virtual Auditorium, or to call another person directly. l Using Infinity Connect to share your screen. l Using the Pexip Mobile App to control the conference. © 2014 Pexip AS Version 8.a December 2014 Page 1 of 18 Pexip Infinity User Guide Using your Virtual Meeting Room Using your Virtual Meeting Room About your Virtual Meeting Room Your Virtual Meeting Room (VMR) is a personal space that is always available for you to meet with others over video or audio, and share presentations. It has a unique alias that you and the other participants dial to join the conference, and it can optionally be PIN-protected. Joining the conference You do not need to specially reserve the VMR or schedule a time for the conference because the VMR is always available for your personal use. When you and the other participants want to meet in your Virtual Meeting Room: 1. From and Infinity Connect client or your preferred video endpoints, each dial the conference alias. 2. If prompted for a PIN, you should enter the host PIN.