Viral Genomics in Ebola Virus Research
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Multicenter, Multi-Outbreak, Randomized, Controlled Safety And
2018 Ebola MCM RCT Protocol Page 1 of 92 IND#: 125530 CONFIDENTIAL 4 October 2019, version 7.0 A Multicenter, Multi-Outbreak, Randomized, Controlled Safety and Efficacy Study of Investigational Therapeutics for the Treatment of Patients with Ebola Virus Disease Short Title: 2018 Ebola MCM RCT Protocol Sponsored by: Office of Clinical Research Policy and Regulatory Operations (OCRPRO) National Institute of Allergy and Infectious Diseases 5601 Fishers Lane Bethesda, MD 20892 NIH Protocol Number: 19-I-0003 Protocol IND #: 125530 ClinicalTrials.gov Number: NCT03719586 Date: 4 October 2019 Version: 7.0 CONFIDENTIAL This document is confidential. No part of it may be transmitted, reproduced, published, or used by other persons without prior written authorization from the study sponsor and principal investigator. 2018 Ebola MCM RCT Protocol Page 2 of 92 IND#: 125530 CONFIDENTIAL 4 October 2019, version 7.0 KEY ROLES DRC Principal Investigator: Jean-Jacques Muyembe-Tamfum, MD, PhD Director-General, DRC National Institute for Biomedical Research Professor of Microbiology, Kinshasa University Medical School Kinshasa Gombe Democratic Republic of the Congo Phone: +243 898949289 Email: [email protected] Other International Investigators: see Appendix E Statistical Lead: Lori Dodd, PhD Biostatistics Research Branch, DCR, NIAID 5601 Fishers Lane, Room 4C31 Rockville, MD 20852 Phone: 240-669-5247 Email: [email protected] U.S. Principal Investigator: Richard T. Davey, Jr., MD Clinical Research Section, LIR, NIAID, NIH Building 10, Room 4-1479, Bethesda, -

Fighting Ebola: Voices from the Frontline Documenting the Experiences of East African Deployed Experts Who Volunteered to Fight Ebola in West Africa Implemented By
Fighting Ebola: Voices from the Frontline Documenting the experiences of East African deployed experts who volunteered to fight Ebola in West Africa Implemented by: Fighting Ebola: Voices from the Frontline Documenting the experiences of East African deployed experts who volunteered to fight Ebola in West Africa Contents Introduction . 6 Foreword from the Hon . Jesca Eriyo . 9 Foreword from Dr Zabulon Yoti . .. 10 Foreword from Dr Jackson Amone . 12 Foreword from Dr Irene Lukassowitz . 13 George Acire . 14 Rebecca Racheal Apolot . 17 Sarah Awilo . 22 Dr Mwaniki Collins . 24 Charles Draleku . 26 Emmanuel Ejoku .. 28 Dr Madina Hussein . 31 Loveness Daniel Isojick . 34 Dr Abdulrahman Said Kassim . 36 Teddy Kusemererwa . .. 40 Liliane Luwaga . 43 Dr Appolinaire Manirafasha . 46 Dr Landry Mayigane . 48 James Mugume . 50 Dr Monica Musenero . 52 Doreen Nabawanuka . 55 Dr Bella Nihorimbere . 56 Teresia Wairimu Thuku . 57 Tony Walter Onena . 59 Acknowledgements . 61 4 Fighting Ebola: Voices from the Frontline 5 Introduction ccording to the World Health Organization (WHO), Others, seeing scenes of death and devastation on their The East African Community Secretariat, in collaboration the Ebola epidemic that occurred in West Africa television screens, just felt compelled to do whatever they with the Federal Government of Germany through the Abetween 2014 and 2016 killed over 11,000 people could to help . Given that so many West African health GIZ-coordinated Support to Pandemic Preparedness in out of the almost 30,000 that were infected . From one -

Uveal Involvement in Marburg Virus Disease B
Br J Ophthalmol: first published as 10.1136/bjo.61.4.265 on 1 April 1977. Downloaded from British Journal of Ophthalmology, 1977, 61, 265-266 Uveal involvement in Marburg virus disease B. S. KUMING AND N. KOKORIS From the Department of Ophthalmology, Johannesburg General Hospital and University of the Witwatersrand SUMMARY The first reported case of uveal involvement in Marburg virus disease is described. 'Ex Africa semper aliquid novi'. Two outbreaks of Marburg virus disease have been Rhodesia and had also been constantly at his documented. The first occurred in Marburg and bedside till his death. Lassa fever was suspected and Frankfurt, West Germany, in 1967 (Martini, 1969) she was given a unit of Lassa fever convalescent and the second in Johannesburg in 1975 (Gear, serum when she became desperately ill on the fifth 1975). This case report describes the third patient day. She also developed acute pancreatitis. Within in the Johannesburg outbreak, who developed an 52 hours she made a dramatic and uneventful anterior uveitis. The cause of the uveitis was proved recovery. Her illness mainly affected the haema- to be the Marburg virus by identiying it in a tissue topoietic, hepatic, and pancreatic systems. culture of her aqueous fluid. The subject of this report was a nurse who had helped to nurse patients 1 and 2. Nine days after the Case report death ofthe first patient she presented with lower back pain and high fever. She developed hepatitis, a mild Before describing the case history of the patient the disseminated intravascular coagulation syndrome, events leading to her contracting the disease must successfully treated with heparin, and the classical be briefly described. -

Ebola Virus Disease
Outbreaks Chronology: Ebola Virus Disease Known Cases and Outbreaks of Ebola Virus Disease, in Reverse Chronological Order: Reported number (%) of Reported deaths Ebola number of among Year(s) Country subtype human cases cases Situation August- Democratic Ebola virus 66 49 (74%) Outbreak occurred in November 2014 Republic of multiple villages in the Congo the Democratic Republic of the Congo. The outbreak was unrelated to the outbreak of Ebola in West Africa. March 2014- Multiple Ebola virus 28652 11325 Outbreak across Present countries multiple countries in West Africa. Number of patients is constantly evolving due to the ongoing investigation. 32 November 2012- Uganda Sudan virus 6* 3* (50%) Outbreak occurred in January 2013 the Luwero District. CDC assisted the Ministry of Health in the epidemiologic and diagnostic aspects of the outbreak. Testing of samples by CDC's Viral Special Pathogens Branch occurred at UVRI in Entebbe. 31 June-November Democratic Bundibugyo 36* 13* (36.1%) Outbreak occurred in 2012 Republic of virus DRC’s Province the Congo Orientale. Laboratory support was provided through CDC and the Public Health Agency of Canada (PHAC)’s field laboratory in Isiro, as well as through the CDC/UVRI lab in Uganda. The outbreak in DRC had no epidemiologic link to the near contemporaneous Ebola outbreak in the Kibaale district of Uganda. 31 June-October Uganda Sudan virus 11* 4* (36.4%) Outbreak occurred in 2012 the Kibaale District of Uganda. Laboratory tests of blood samples were conducted by the UVRI and the CDC. 31 May 2011 Uganda Sudan virus 1 1 (100%) The Uganda Ministry of Health informed the public a patient with suspected Ebola Hemorrhagic fever died on May 6, 2011 in the Luwero district, Uganda. -

Targeting Ebola 2015
Agenda of Targeting Ebola 2015 May 28-29, 2015 Institut Pasteur - Paris, France www.targeting-ebola.com Welcome Note On behalf of the Targeting Infectious Diseases Committee, the Institut Pasteur, the COPED and the Task Force for Ebola in France, it is our pleasure to announce the organization of the International Congress on Targeting Ebola 2015 which will be held on May 28-29, 2015 at Pasteur Institute, Paris, France. The World Health Organization (WHO) was notified on March 23, 2014, of an outbreak of EVD in Guinea. The disease soon spread to the bordering countries of Liberia and Sierra Leone, which are the most severely affected countries. On August 8, 2014, the epidemic was declared a “public health emergency of international concern” (WHO Ebola Response Team, NEJM 2014, 371, 1481). Suspected cases of EVD have since been reported in seven affected countries (Guinea, Liberia, Nigeria, Senegal, Sierra Leone, Spain, and the United States of America). Unprecedented in scale and geographical distribution since the identification of Ebola in 1976, the current epidemic has an apparent overall case-fatality ratio of about 70%; but it is suspected that many more cases have gone unrecorded. On May 2015, more than 10.000 deaths and 30.000 cases had been reported in Sierra Leone, Liberia and Guinea, according to the WHO. Globally, the situation is improving. The epidemic is over in Liberia, however in Guinea and Sierra Leone, the initial decline observed a few weeks ago is stable with approximately 20 new cases / week. The vast majority of cases are reported from the western prefecture of Forecariah, which borders the Sierra Leonean district of Kambia. -

Clinical Trial Protocol Sponsored by OCRPRO
PREVAIL IV: Double-blind, Randomized, Two-phase, Placebo-controlled, Phase II Trial of GS-5734 to Assess the Antiviral Activity, Longer-term Clearance of Ebola Virus, and Safety in Male Ebola Survivors with Evidence of Ebola Virus Persistence in Semen NIAID Protocol Number: 16-I-N137 Version Number: 10.0 Date: May 13, 2019 Investigational New Drug (IND) Number: 130621 IND Sponsor: Office of Clinical Research Policy and Regulatory Operations (OCRPRO), Division of Clinical Research (DCR), National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH) Local Liberian Medical Officer: Ian Wachekwa, MD Safety Medical Officer John F. Kennedy Hospital 21 Street, Sinkor Monrovia, Liberia [email protected] (t) +231880890907 Sponsor Medical Monitor: Alan Lifson, MD, MPH Professor, Division of Epidemiology and Community Health School of Public Health University of Minnesota 1300 S. Second St., Ste. 300 Minneapolis, MN 55454 (t) 612-626-9697 (f) 612-624-0315 [email protected] Pharmaceutical Support Provided by: Gilead Sciences, Inc. PREVAIL IV Version 10.0 13 May 2019 Conducted by: Liberia-US Joint Clinical Research Partnership also known as the Partnership for Research on Ebola Virus in Liberia (PREVAIL) Page 2 of 77 PREVAIL IV Version 10.0 13 May 2019 Key Roles NIH Principal Investigator: Elizabeth S. Higgs, MD, MIA, DTMH Division of Clinical Research, NIAID [email protected] (c) +301-768-3947 skype: libby.higgs2 Liberian Principal Investigator: Dehkontee Gayedyu-Dennis, MD PREVAIL [email protected] (t) +231-886-538-810 -
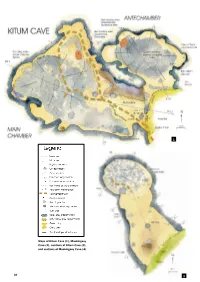
30 SWARA October – December 2007 Maps of Kitum Cave
1 Maps of Kitum Cave (1), Mackingeny Cave (2), sections of Kitum Cave (3), and sections of Mackingeny Cave (4). 30 SWARA October – December 2007 2 behaviour 3 Mount Elgon’s ‘elephant caves’ Joyce Lundberg and Donald McFarlane contemplate and map the extraordinary underground attractions of western Kenya’s Mount Elgon National Park. 4 SWARA October – December 2007 31 ne of Kenya’s least vis- that are far too busy eating the herbivore ited parks, the Mount equivalent of chocolate mousse to take Elgon National Park much notice of their potential fate. Oprotects a narrow band The salts are mainly mirabalite, of forest climbing the eastern flank sodium sulphate (commonly called of East Africa’s fifth highest mas- Glauber’s salt), which grows out sif, standing 4,321 metres (14,178 from the walls and can form curved crys- feet) above sea level (see map below). tals resembling pig’s tusks. The crystals are For most visitors, the Park’s principal licked off the wall by buffalos or scraped/ attraction rests in its suite of unique caves, gouged out by bushbuck and elephants. of which the Kitum and the Makingeny Buffalos cannot scrape the rock themselves, Caves are best known. Used by the Elgony so they eat mainly the leftover bits dropped people for centuries, the Elgon caves came by the elephants. The marks left by bush- to wider attention through the writings of buck teeth and elephant tusks are clearly Joseph Thomson (Through Masai Land, identifiable on the cave walls. 1885), and are thought to have been the These marks should not be confused inspiration too for H. -

As a Model of Human Ebola Virus Infection
Viruses 2012, 4, 2400-2416; doi:10.3390/v4102400 OPEN ACCESS viruses ISSN 1999-4915 www.mdpi.com/journal/viruses Review The Baboon (Papio spp.) as a Model of Human Ebola Virus Infection Donna L. Perry 1,*, Laura Bollinger 1 and Gary L.White 2 1 Integrated Research Facility, Division of Clinical Research, NIAID, NIH, Frederick, MD, USA; E-Mail: [email protected] 2 Department of Pathology, University of Oklahoma Baboon Research Resource, University of Oklahoma, Ft. Reno Science Park, OK, USA; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-301-631-7249; Fax: +1-301-619-5029. Received: 8 October 2012; in revised form: 17 October 2012 / Accepted: 17 October 2012 / Published: 23 October 2012 Abstract: Baboons are susceptible to natural Ebola virus (EBOV) infection and share 96% genetic homology with humans. Despite these characteristics, baboons have rarely been utilized as experimental models of human EBOV infection to evaluate the efficacy of prophylactics and therapeutics in the United States. This review will summarize what is known about the pathogenesis of EBOV infection in baboons compared to EBOV infection in humans and other Old World nonhuman primates. In addition, we will discuss how closely the baboon model recapitulates human EBOV infection. We will also review some of the housing requirements and behavioral attributes of baboons compared to other Old World nonhuman primates. Due to the lack of data available on the pathogenesis of Marburg virus (MARV) infection in baboons, discussion of the pathogenesis of MARV infection in baboons will be limited. -

Replication of Marburg Virus in Human Endothelial Cells a Possible Mechanism for the Development of Viral Hemorrhagic Disease
Replication of Marburg Virus in Human Endothelial Cells A Possible Mechanism for the Development of Viral Hemorrhagic Disease Hans-Joachim Schnittler,$ Friederike Mahner, * Detlev Drenckhahn, * Hans-Dieter Klenk, * and Heinz Feldmann * *Institut fir Virologie, Philipps-Universitdt Marburg, 3550 Marburg, Germany; tInstitutftirAnatomie, Universitdt Wiirzburg, 8700 Wiirzburg, Germany Abstract Rhabdoviridae, within the new proposed order Mononegavi- Marburg and Ebola virus, members of the family Filoviridae, rales (10). Virions are composed of a helical nucleocapsid cause a severe hemorrhagic disease in humans and primates. surrounded by a lipid envelope. The genome is nonsegmented, The disease is characterized as a pantropic virus infection often of negative sense, and 19 kb in length (3, 11, 12). Virion parti- resulting in a fulminating shock associated with hemorrhage, cles contain at least seven structural proteins (8, 13-16). and death. All known histological and pathophysiological pa- Filovirus infections have several pathological features in rameters of the disease are not sufficient to explain the devas- common with other severe viral hemorrhagic fevers such as tating symptoms. Previous studies suggested a nonspecific de- Lassa fever, hemorrhagic fever with renal syndrome, and struction of the endothelium as a possible mechanism. Con- Dengue hemorrhagic fever (5). Among these viruses, filovi- cerning the important regulatory functions of the endothelium ruses cause the highest case-fatality rates ( - 35% for MBG [61 (blood pressure, antithrombogenicity, homeostasis), we exam- and up to 90% for EBO, subtype Zaire [17]) and the most ined Marburg virus replication in primary cultures of human severe hemorrhagic manifestations. The pathophysiologic endothelial cells and organ cultures of human umbilical cord events that make filovirus infections of humans so devastating veins. -

Rabbit Anti-Marburgvirus (MARV) VLP Pab ELISA Data
4 Research Court, Suite 300 Rockville, MD 20850 877-411-2041 [email protected] Rabbit anti-Marburgvirus (MARV) VLP pAb ELISA Data: Catalog #: 04-0005 IgG IgG + IgG + Lot #: MMIG201001IBT Dilution MMARV ZEBOV 1:X Antigen Antigen Immunogen: MARV (Musoke strain) Virus-like Particles (VLPs) containing glycoprotein (GP) 1000 3.30 2.66 Nucleoprotein (NP), and viral protein (VP40). 3162 3.07 1.80 10000 2.68 0.89 Description: Protein A purified rabbit polyclonal 31623 1.87 0.37 antibody reactive to MARV VLP raised in New 100000 0.99 0.14 Zealand white rabbits. 316228 0.43 0.05 1000000 0.17 0.02 Supplied: 0.5 mg of antibody is supplied in PBS at a concentration of 5.75 mg/mL. 0.01% Sodium azide has been added. -Antigen is coated on ELISA plates overnight. -Add 200µl blocking buffer then wash wells with Clonality: Polyclonal PBST. -Antiserum is diluted semi-log. Relevance: the filovirus Marburgvirus is a -Incubate antibody for 2 hour. Category A (NIAID) and HHS select agent. -Wash unbound antibodies and add HRP- Recommended Dilutions: conjugated anti-rabbit IgG. -Wash plates and add substrate to develop color for ELISA: Assay-dependent dilution. 20 minutes. WB: Assay-dependent dilution -Read absorbance at 650nm. Amount of color is directly proportional to amount of antibodies. Storage: 2-3 weeks +4oC, -20◦C long term Western Blot Cross Reactivity: Historical data showed some cross-reactivity with Ebola Virus (EBOV) and -Antiserum recognizes Marburg musoke Sudan Virus (SUDV) VLP’s, most likely due to glycoprotein, nucleoprotein, and VP40 antibodies against Baculovirus proteins since the VLP’s were expressed in SF9-Baculovirus system. -

Comparison and Evaluation of Real-Time Taqman PCR for Detection and Quantification of Ebolavirus
viruses Article Comparison and Evaluation of Real-Time Taqman PCR for Detection and Quantification of Ebolavirus Yi Huang 1,* , Shuqi Xiao 2 and Zhiming Yuan 1,* 1 National Biosafety Laboratory, Chinese Academy of Sciences, Wuhan 430020, China 2 Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan 430020, China; [email protected] * Correspondence: [email protected] (Y.H.); [email protected] (Z.Y.) Abstract: Given that ebolavirus causes severe and frequently lethal disease, its rapid and accurate detection using available and validated methods is essential for controlling infection. Real-time reverse-transcription PCR (RT-PCR) has proven to be an invaluable tool for ebolaviruses diagnostics. Many assays with different targets have been developed, but they have not been externally compared or validated, and limits of detection are not uniformly reported. Here we compared and evaluated the sensitivity, reproducibility and specificity of 23 in-house assays under the same conditions. Our results showed that these assays were highly gene- and species- specific when evaluated using in vitro RNA transcripts and viral RNA, and the potential limits of detection were uniformly reported 2 6 ranging from 10 to 10 in vitro synthesized RNA transcripts copies perµL and 1–100 TCID50/mL. The comparison of these assays indicated that those targeting the more conservative NP gene could be the better option for EVD case definition and quantitative measurement because of its higher sensitivity for the same species. Our analysis could contribute to the standardization of ebolavirus detection and quantification assays, which can offer a better understanding of the meaning of results across laboratories and time points, as well as make them easy to implement, especially under outbreak conditions. -

The Hemorrhagic Fevers of Southern Africa South African Institute For
THE YALE JOURNAL OF BIOLOGY AND MEDICINE 55 (1982), 207-212 The Hemorrhagic Fevers of Southern Africa with Special Reference to Studies in the South African Institute for Medical Research J.H.S. GEAR, M.D. National Institute for Virology, Johannesburg, South Africa Received April 19, 1982 In this review of studies on the hemorrhagic fevers of Southern Africa carried out in the South African Institute for Medical Research, attention has been called to occurrence of meningococcal septicemia in recruits to the mining industry and South African Army, to cases of staphylococcal and streptococcal septicemia with hemorrhagic manifestations, and to the occurrence of plague which, in its septicemic form, may cause a hemorrhagic state. "Onyalai," a bleeding disease in tropical Africa, often fatal, was related to profound throm- bocytopenia possibly following administration of toxic witch doctor medicine. Spirochetal diseases, and rickettsial diseases in their severe forms, are often manifested with hemorrhagic complications. Of enterovirus infections, Coxsackie B viruses occasionally caused severe hepa- titis associated with bleeding, especially in newborn babies. Cases of hemorrhagic fever presenting in February-March, 1975 are described. The first out- break was due to Marburg virus disease and the second, which included seven fatal cases, was caused by Rift Valley fever virus. In recent cases of hemorrhagic fever a variety of infective organisms have been incriminated including bacterial infections, rickettsial diseases, and virus diseases, including Herpesvirus hominis; in one patient, the hemorrhagic state was related to rubella. A boy who died in a hemorrhagic state was found to have Congo fever; another pa- tient who died of severe bleeding from the lungs was infected with Leptospira canicola, and two patients who developed a hemorrhagic state after a safari trip in Northern Botswana were infected with Trypanosoma rhodesiense.