Rsubread/Subread Users Guide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

At—At, Batch—Execute Commands at a Later Time
at—at, batch—execute commands at a later time at [–csm] [–f script] [–qqueue] time [date] [+ increment] at –l [ job...] at –r job... batch at and batch read commands from standard input to be executed at a later time. at allows you to specify when the commands should be executed, while jobs queued with batch will execute when system load level permits. Executes commands read from stdin or a file at some later time. Unless redirected, the output is mailed to the user. Example A.1 1 at 6:30am Dec 12 < program 2 at noon tomorrow < program 3 at 1945 pm August 9 < program 4 at now + 3 hours < program 5 at 8:30am Jan 4 < program 6 at -r 83883555320.a EXPLANATION 1. At 6:30 in the morning on December 12th, start the job. 2. At noon tomorrow start the job. 3. At 7:45 in the evening on August 9th, start the job. 4. In three hours start the job. 5. At 8:30 in the morning of January 4th, start the job. 6. Removes previously scheduled job 83883555320.a. awk—pattern scanning and processing language awk [ –fprogram–file ] [ –Fc ] [ prog ] [ parameters ] [ filename...] awk scans each input filename for lines that match any of a set of patterns specified in prog. Example A.2 1 awk '{print $1, $2}' file 2 awk '/John/{print $3, $4}' file 3 awk -F: '{print $3}' /etc/passwd 4 date | awk '{print $6}' EXPLANATION 1. Prints the first two fields of file where fields are separated by whitespace. 2. Prints fields 3 and 4 if the pattern John is found. -

Unix Introduction
Unix introduction Mikhail Dozmorov Summer 2018 Mikhail Dozmorov Unix introduction Summer 2018 1 / 37 What is Unix Unix is a family of operating systems and environments that exploits the power of linguistic abstractions to perform tasks Unix is not an acronym; it is a pun on “Multics”. Multics was a large multi-user operating system that was being developed at Bell Labs shortly before Unix was created in the early ’70s. Brian Kernighan is credited with the name. All computational genomics is done in Unix http://www.read.seas.harvard.edu/~kohler/class/aosref/ritchie84evolution.pdfMikhail Dozmorov Unix introduction Summer 2018 2 / 37 History of Unix Initial file system, command interpreter (shell), and process management started by Ken Thompson File system and further development from Dennis Ritchie, as well as Doug McIlroy and Joe Ossanna Vast array of simple, dependable tools that each do one simple task Ken Thompson (sitting) and Dennis Ritchie working together at a PDP-11 Mikhail Dozmorov Unix introduction Summer 2018 3 / 37 Philosophy of Unix Vast array of simple, dependable tools Each do one simple task, and do it really well By combining these tools, one can conduct rather sophisticated analyses The Linux help philosophy: “RTFM” (Read the Fine Manual) Mikhail Dozmorov Unix introduction Summer 2018 4 / 37 Know your Unix Unix users spend a lot of time at the command line In Unix, a word is worth a thousand mouse clicks Mikhail Dozmorov Unix introduction Summer 2018 5 / 37 Unix systems Three common types of laptop/desktop operating systems: Windows, Mac, Linux. Mac and Linux are both Unix-like! What that means for us: Unix-like operating systems are equipped with “shells”" that provide a command line user interface. -
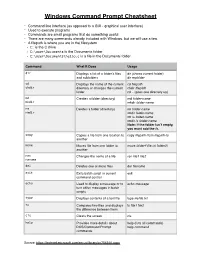
Windows Command Prompt Cheatsheet
Windows Command Prompt Cheatsheet - Command line interface (as opposed to a GUI - graphical user interface) - Used to execute programs - Commands are small programs that do something useful - There are many commands already included with Windows, but we will use a few. - A filepath is where you are in the filesystem • C: is the C drive • C:\user\Documents is the Documents folder • C:\user\Documents\hello.c is a file in the Documents folder Command What it Does Usage dir Displays a list of a folder’s files dir (shows current folder) and subfolders dir myfolder cd Displays the name of the current cd filepath chdir directory or changes the current chdir filepath folder. cd .. (goes one directory up) md Creates a folder (directory) md folder-name mkdir mkdir folder-name rm Deletes a folder (directory) rm folder-name rmdir rmdir folder-name rm /s folder-name rmdir /s folder-name Note: if the folder isn’t empty, you must add the /s. copy Copies a file from one location to copy filepath-from filepath-to another move Moves file from one folder to move folder1\file.txt folder2\ another ren Changes the name of a file ren file1 file2 rename del Deletes one or more files del filename exit Exits batch script or current exit command control echo Used to display a message or to echo message turn off/on messages in batch scripts type Displays contents of a text file type myfile.txt fc Compares two files and displays fc file1 file2 the difference between them cls Clears the screen cls help Provides more details about help (lists all commands) DOS/Command Prompt help command commands Source: https://technet.microsoft.com/en-us/library/cc754340.aspx. -

Creating Rpms Guide
CREATING RPMS (Student version) v1.0 Featuring 36 pages of lecture and a 48 page lab exercise This docu m e n t serves two purpose s: 1. Representative sample to allow evaluation of our courseware manuals 2. Make available high quality RPM documentation to Linux administrators A bout this m aterial : The blue background you see simulates the custom paper that all Guru Labs course w are is printed on. This student version does not contain the instructor notes and teaching tips present in the instructor version. For more information on all the features of our unique layout, see: http://ww w . g urulabs.co m /courseware/course w are_layout.php For more freely available Guru Labs content (and the latest version of this file), see: http://www.gurulabs.co m/goodies/ This sample validated on: Red Hat Enterprise Linux 4 & Fedora Core v3 SUSE Linux Enterprise Server 9 & SUSE Linux Professional 9.2 About Guru Labs: Guru Labs is a Linux training company started in 199 9 by Linux experts to produce the best Linux training and course w are available. For a complete list, visit our website at: http://www.gurulabs.co m/ This work is copyrighted Guru Labs, L.C. 2005 and is licensed under the Creative Common s Attribution- NonCom mer cial- NoDerivs License. To view a copy of this license, visit http://creativecom m o n s.org/licenses/by- nc- nd/2.0/ or send a letter to Creative Commons, 559 Nathan Abbott Way, Stanford, California 943 0 5, USA. Guru Labs 801 N 500 W Ste 202 Bountiful, UT 84010 Ph: 801-298-5227 WWW.GURULABS.COM Objectives: • Understand -

The Frege Programming Language ( Draft)
The Frege Programming Language (Draft) by Ingo Wechsung last changed May 14, 2014 3.21.285 Abstract This document describes the functional programming language Frege and its implemen- tation for the Java virtual machine. Commonplace features of Frege are type inference, lazy evaluation, modularization and separate compile-ability, algebraic data types and type classes, pattern matching and list comprehension. Distinctive features are, first, that the type system supports higher ranked polymorphic types, and, second, that Frege code is compiled to Java. This allows for maximal interoperability with existing Java software. Any Java class may be used as an abstract data type, Java functions and methods may be called from Frege functions and vice versa. Despite this interoperability feature Frege is a pure functional language as long as impure Java functions are declared accordingly. What is or who was Frege? Friedrich Ludwig Gottlob Frege was a German mathematician, who, in the second half of the 19th century tried to establish the foundation of mathematics in pure logic. Al- though this attempt failed in the very moment when he was about to publish his book Grundgesetze der Arithmetik, he is nevertheless recognized as the father of modern logic among philosophers and mathematicians. In his essay Funktion und Begriff [1] Frege introduces a function that takes another function as argument and remarks: Eine solche Funktion ist offenbar grundverschieden von den bisher betrachteten; denn als ihr Argument kann nur eine Funktion auftreten. Wie nun Funktionen von Gegenst¨andengrundverschieden sind, so sind auch Funktionen, deren Argu- mente Funktionen sind und sein m¨ussen,grundverschieden von Funktionen, deren Argumente Gegenst¨andesind und nichts anderes sein k¨onnen. -

Praat Scripting Tutorial
Praat Scripting Tutorial Eleanor Chodroff Newcastle University July 2019 Praat Acoustic analysis program Best known for its ability to: Visualize, label, and segment audio files Perform spectral and temporal analyses Synthesize and manipulate speech Praat Scripting Praat: not only a program, but also a language Why do I want to know Praat the language? AUTOMATE ALL THE THINGS Praat Scripting Why can’t I just modify others’ scripts? Honestly: power, flexibility, control Insert: all the gifs of ‘you can do it’ and ‘you got this’ and thumbs up Praat Scripting Goals ~*~Script first for yourself, then for others~*~ • Write Praat scripts quickly, effectively, and “from scratch” • Learn syntax and structure of the language • Handle various input/output combinations Tutorial Overview 1) Praat: Big Picture 2) Getting started 3) Basic syntax 4) Script types + Practice • Wav files • Measurements • TextGrids • Other? Praat: Big Picture 1) Similar to other languages you may (or may not) have used before • String and numeric variables • For-loops, if else statements, while loops • Regular expression matching • Interpreted language (not compiled) Praat: Big Picture 2) Almost everything is a mouse click! i.e., Praat is a GUI scripting language GUI = Graphical User Interface, i.e., the Objects window If you ever get lost while writing a Praat script, click through the steps using the GUI Getting Started Open a Praat script From the toolbar, select Praat à New Praat script Save immediately! Save frequently! Script Goals and Input/Output • Consider what -

Deviceinstaller User Guide
Device Installer User Guide Part Number 900-325 Revision C 03/18 Table of Contents 1. Overview ...................................................................................................................................... 1 2. Devices ........................................................................................................................................ 2 Choose the Network Adapter for Communication ....................................................................... 2 Search for All Devices on the Network ........................................................................................ 2 Change Views .............................................................................................................................. 2 Add a Device to the List ............................................................................................................... 3 View Device Details ..................................................................................................................... 3 Device Lists ................................................................................................................................. 3 Save the Device List ................................................................................................................ 3 Open the Device List ............................................................................................................... 4 Print the Device List ................................................................................................................ -

Sys, Shutil, and OS Modules
This video will introduce the sys, shutil, glob, and os modules. 1 The sys module contains a few tools that are often useful and will be used in this course. The first tool is the argv function which allows the script to request a parameter when the script is run. We will use this function in a few weeks when importing scripts into ArcGIS. If a zero is specified for the argv function, then it will return the location of the script on the computer. This can be helpful if the script needs to look for a supplemental file, such as a template, that is located in the same folder as the script. The exit function stops the script when it reach the exit statement. The function can be used to stop a script when a certain condition occurs – such as when a user specifies an invalid parameter. This allows the script to stop gracefully rather than crashing. The exit function is also useful when troubleshooting a script. 2 The shutil module contains tools for moving and deleting files. The copy tool makes a copy of a file in a new location. The copytree tool makes a copy of an entire folder in a new location. The rmtree tool deletes an entire folder. Note that this tool will not send items to the recycling bin and it will not ask for confirmation. The tool will fail if it encounters any file locks in the folder. 3 The os module contains operating system functions – many of which are often useful in a script. -

Lenovo Flex System FC3171 8 Gb SAN Switch Command Line Interface User’S Guide This Document Explains How to Manage the Switch Using the CLI
Flex System FC3171 8 Gb SAN Switch Command Line Interface User’s Guide Flex System FC3171 8 Gb SAN Switch Command Line Interface User’s Guide Note: Before using this information and the product it supports, read the general information in “Notices” on page 391. First Edition, April 2015 © Copyright Lenovo 2015. LIMITED AND RESTRICTED RIGHTS NOTICE: If data or software is delivered pursuant a General Services Administration “GSA” contract, use, reproduction, or disclosure is subject to restrictions set forth in Contract No. GS-35F-05925. Contents Chapter 1. Lenovo Flex System FC3171 8 Gb SAN Switch . 1 Related documentation . 1 Notices and statements in this document . 3 Chapter 2. Command line interface usage . 5 Logging in to the switch . 6 Opening and closing an Admin session . 7 Entering commands. 7 Getting help . 7 Setting page breaks . 8 Creating a support file. 9 Downloading and uploading files. 10 Chapter 3. User account configuration . 13 Displaying user account information . 14 Creating user accounts . 15 Modifying user accounts and passwords. 15 Chapter 4. Network and fabric configuration . 17 Displaying the Ethernet network configuration . 17 Displaying name server information . 18 Configuring the Ethernet port . 19 IPv4 configuration . 19 IPv6 configuration . 20 DNS server configuration . 21 Verifying a switch in the network . 22 Verifying and tracing Fibre Channel connections . 22 Managing IP security. 23 IP security concepts . 24 Legacy and Strict security . 24 Security policies and associations . 24 IKE peers and policies . 25 Public key infrastructure . 25 Displaying IP security information . 25 Policy and association information . 25 IKE peer and policy information . 26 Public key infrastructure information . -

PS TEXT EDIT Reference Manual Is Designed to Give You a Complete Is About Overview of TEDIT
Information Management Technology Library PS TEXT EDIT™ Reference Manual Abstract This manual describes PS TEXT EDIT, a multi-screen block mode text editor. It provides a complete overview of the product and instructions for using each command. Part Number 058059 Tandem Computers Incorporated Document History Edition Part Number Product Version OS Version Date First Edition 82550 A00 TEDIT B20 GUARDIAN 90 B20 October 1985 (Preliminary) Second Edition 82550 B00 TEDIT B30 GUARDIAN 90 B30 April 1986 Update 1 82242 TEDIT C00 GUARDIAN 90 C00 November 1987 Third Edition 058059 TEDIT C00 GUARDIAN 90 C00 July 1991 Note The second edition of this manual was reformatted in July 1991; no changes were made to the manual’s content at that time. New editions incorporate any updates issued since the previous edition. Copyright All rights reserved. No part of this document may be reproduced in any form, including photocopying or translation to another language, without the prior written consent of Tandem Computers Incorporated. Copyright 1991 Tandem Computers Incorporated. Contents What This Book Is About xvii Who Should Use This Book xvii How to Use This Book xvii Where to Go for More Information xix What’s New in This Update xx Section 1 Introduction to TEDIT What Is PS TEXT EDIT? 1-1 TEDIT Features 1-1 TEDIT Commands 1-2 Using TEDIT Commands 1-3 Terminals and TEDIT 1-3 Starting TEDIT 1-4 Section 2 TEDIT Topics Overview 2-1 Understanding Syntax 2-2 Note About the Examples in This Book 2-3 BALANCED-EXPRESSION 2-5 CHARACTER 2-9 058059 Tandem Computers -

RL78/G23 Unique ID Read Driver Introduction Each RL78/G2x Chip Is Programmed with a Unique ID
Application Note RL78/G23 Unique ID Read Driver Introduction Each RL78/G2x chip is programmed with a unique ID. The unique ID can be used to prevent unauthorized use of software IP and is useful for managing products individually. This application note presents unique ID usage examples and describes how to use the unique ID read driver. The driver reads the 16-byte unique ID and 9-byte product name stored as ASCII code in the extra area and writes them to a specified area. Target Device RL78/G23 When using this application note with other Renesas MCUs, careful evaluation is recommended after making modifications to comply with the alternate MCU. R20AN0615EJ0100 Rev.1.00 Page 1 of 14 Apr.13.21 RL78/G23 Unique ID Read Driver Contents 1. Overview .................................................................................................................................... 3 1.1 About This Application Note .................................................................................................................... 3 1.2 Confirmed Operation Environment .......................................................................................................... 3 2. About the Unique ID .................................................................................................................. 5 2.1 Unique ID Specifications ......................................................................................................................... 5 2.2 Unique ID Usage Examples ................................................................................................................... -

ANSWERS ΤΟ EVEN-Numbered
8 Answers to Even-numbered Exercises 2.1. WhatExplain the following unexpected are result: two ways you can execute a shell script when you do not have execute permission for the file containing the script? Can you execute a shell script if you do not have read permission for the file containing the script? You can give the name of the file containing the script as an argument to the shell (for example, bash scriptfile or tcsh scriptfile, where scriptfile is the name of the file containing the script). Under bash you can give the following command: $ . scriptfile Under both bash and tcsh you can use this command: $ source scriptfile Because the shell must read the commands from the file containing a shell script before it can execute the commands, you must have read permission for the file to execute a shell script. 4.3. AssumeWhat is the purpose ble? you have made the following assignment: $ person=zach Give the output of each of the following commands. a. echo $person zach b. echo '$person' $person c. echo "$person" zach 1 2 6.5. Assumengs. the /home/zach/grants/biblios and /home/zach/biblios directories exist. Specify Zach’s working directory after he executes each sequence of commands. Explain what happens in each case. a. $ pwd /home/zach/grants $ CDPATH=$(pwd) $ cd $ cd biblios After executing the preceding commands, Zach’s working directory is /home/zach/grants/biblios. When CDPATH is set and the working directory is not specified in CDPATH, cd searches the working directory only after it searches the directories specified by CDPATH.