C 2016 by Henry A. Lin. All Rights Reserved
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Marriott's Oceana Palms |Singer Island, Riviera Beach, Florida
Marriott’s Oceana Palms | Singer Island, Riviera Beach, Florida Week 2020 Check-In Days 2021 Check-In Days Season No. Thursday Friday Saturday Sunday Thursday Friday Saturday Sunday 1 Jan 2 – Jan 9 Jan 3 – Jan 10 Jan 4 – Jan 11 Jan 5 – Jan 12 Jan 7 – Jan 14 Jan 1 – Jan 8 Jan 2 – Jan 9 Jan 3 – Jan 10 2 Jan 9 – Jan 16 Jan 10 – Jan 17 Jan 11 – Jan 18 Jan 12 – Jan 19 Jan 14 – Jan 21 Jan 8 – Jan 15 Jan 9 – Jan 16 Jan 10 – Jan 17 3 Jan 16 – Jan 23 Jan 17 – Jan 24 Jan 18 – Jan 25 Jan 19 – Jan 26 Jan 21 – Jan 28 Jan 15 – Jan 22 Jan 16 – Jan 23 Jan 17 – Jan 24 PLATINUM 4 Jan 23 – Jan 30 Jan 24 – Jan 31 Jan 25 – Feb 1 Jan 26 – Feb 2 Jan 28 – Feb 4 Jan 22 – Jan 29 Jan 23 – Jan 30 Jan 24 – Jan 31 5 Jan 30 – Feb 6 Jan 31 – Feb 7 Feb 1 – Feb 8 Feb 2 – Feb 9 Feb 4 – Feb 11 Jan 29 – Feb 5 Jan 30 – Feb 6 Jan 31 – Feb 7 6 Feb 6 – Feb 13 Feb 7 – Feb 14 Feb 8 – Feb 15 Feb 9 – Feb 16 Feb 11 – Feb 18 Feb 5 – Feb 12 Feb 6 – Feb 13 Feb 7 – Feb 14 PLATINUM PLUS 7 Feb 13 – Feb 20 Feb 14 – Feb 21 Feb 15 – Feb 22 Feb 16 – Feb 23 Feb 18 – Feb 25 Feb 12 – Feb 19 Feb 13 – Feb 20 Feb 14 – Feb 21 PRESIDENTS DAY 8 Feb 20 – Feb 27 Feb 21 – Feb 28 Feb 22 – Feb 29 Feb 23 – Mar 1 Feb 25 – Mar 4 Feb 19 – Feb 26 Feb 20 – Feb 27 Feb 21 – Feb 28 9 Feb 27 – Mar 5 Feb 28 – Mar 6 Feb 29 – Mar 7 Mar 1 – Mar 8 Mar 4 – Mar 11 Feb 26 – Mar 5 Feb 27 – Mar 6 Feb 28 – Mar 7 10 Mar 5 – Mar 12 Mar 6 – Mar 13 Mar 7 – Mar 14 Mar 8 – Mar 15 Mar 11 – Mar 18 Mar 5 – Mar 12 Mar 6 – Mar 13 Mar 7 – Mar 14 11 Mar 12 – Mar 19 Mar 13 – Mar 20 Mar 14 – Mar 21 Mar 15 – Mar 22 Mar 18 – Mar 25 Mar 12 – -
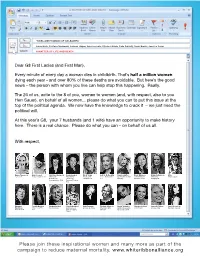
Please Join These Inspirational Women and Many More As Part of the Campaign to Reduce Maternal Mortality
‘WIVES AND HUSBAND OF G8 LEADERS’ Laura Bush, Svetlana Medvedev, Laureen Harper, Veronica Lario, Kiyoko Fukuda, Carla Sarkozy, Sarah Brown, Joachim Sauer A MATTER OF LIFE AND DEATH Dear G8 First Ladies (and First Man), Every minute of every day a woman dies in childbirth. That’s half a million women dying each year - and over 80% of these deaths are avoidable. But here’s the good news – the person with whom you live can help stop this happening. Really. The 26 of us, write to the 8 of you, women to women (and, with respect, also to you Herr Sauer), on behalf of all women... please do what you can to put this issue at the top of the political agenda. We now have the knowledge to crack it – we just need the political will. At this year’s G8, your 7 husbands (and 1 wife) have an opportunity to make history here. There is a real chance. Please do what you can – on behalf of us all. With respect, Emma Thompson Annie Lennox Christiane Amanpour Donna Langley Kirsty Young Lady de Rothschild Claudia Schiffer Wendi Murdoch Elizabeth Edwards Misia Actress, UK Singer, Songwriter, UK CNN Chief President of Broadcaster, CE, LE Rothschild Supermodel, Actress, Chief Strategist, Attourney, Singer, Japan International Production, Presenter, UK LLC Germany Myspace China Campaigner Correspondent, USA Universal Pictures, USA Margaret Fiorella Mannoia Margherita Hack Claudie Haigneré Katja Riemann Elisabeth Murdoch Christy Turlington Christine Ockrent Gwyneth Paltrow Sarah McLachlan MacMillan Singer, Songwriter, Scientist, Italy Astronaut and Actress, Germany Chairman, CEO, CARE Ambassador, COO, France Actress, USA Musician, Canada Historian, USA Italy Advisor to ESA, UK/USA Supermodel, USA France Please join these inspirational women and many more as part of the campaign to reduce maternal mortality. -

Listen to “Circles and Squares”, the Official Song of the ISU World Figure Skating Championships 2021
February 19, 2021 Lausanne, Switzerland Listen to “Circles and Squares”, the Official Song of the ISU World Figure Skating Championships 2021 Today marks the unveiling of “Circles and Squares”, the official song for the 2021 ISU World Figure Skating Championships. The song is performed by international artists Måns Zelmerlöw and Polina Gagarina and was specially commissioned for the event, which will be held at the Ericsson Globe in Stockholm (SWE) from March 22 – 28. The songwriters Måns Zelmerlöw, Stefan Örn, David Lindgren Zacharias, Sandra Bjurman, and Jon Eyden, took their inspiration from Figure Skating. “We wanted to write a song with a slightly serious tone and at the same time bring out the beauty that exists between two Figure Skaters, the passion, the art, and of course mutual respect. This is mirrored in the arrangement of the song that has an intro full of expectation before we bring in the full orchestra that takes the song to the heights we wanted to hit,” says Stefan Örn. They wanted to create a soaring sound, which they did by recording the song in the historical Atlantis Studio. Everything was recorded live, except from the string orchestra, which was recorded by Erik Arwinder. “It’s always fantastic to start with an idea, and then develop it into a full-scale production. Few people have the privilege of working at this level, and I’m humbled to have had this opportunity. Circles and Squares is crafted by a team that I’m proud to work with and right now we’re looking forward to the release of this world-class track!” Örn adds. -

Sister Helen Martinez - Journal for November 17-21, 2014
UN Update - Sister Helen Martinez - Journal for November 17-21, 2014 Saturday morning I went to meet a former colleague from Presentation Elementary- Noreen Greene Fraize, and her daughter, Alanna. Alanna is in her fourth year of studying opera at the Metropolitan School of Music with a focus on Opera. We had brunch at the French Café. I worked all day on Sunday on an analysis response to the Open Working Group proposals for Sustainable Development Goals. My contribution was sent to Elsa who is working with two others. Monday morning, we were fortunate to have some time to hear the experiences of Sister Mary Jo Toll, SNDN, Veronica Brand, RSHM and Justine Gitanjali Senapati, CSJ. They shared their experiences of coming to work at the UN as NGO representatives of their congregations. It was helpful to hear that it takes time to learn how to contribute to various groups according to interests. I feel proud that IPA is part of the UN that influences and impacts systemic change for the world. It fits into how our focus here is on the social protection floor for human rights of people. I learned that NGOs have grown in their influence in draft resolutions by their ability to contact Permanent Missions – in part because religious are involved on the ground level in so many countries. Time here is spent doing research, meeting and participating in working groups, task forces, attending the various sessions at the UN and then sharing this information with those who can influence change. Advocacy work consumes about half of their time. -

Msia Launches Evisa for Chinese Tourists
! PRESS RELEASE FOR IMMEDIATE RELEASE MALAYSIA LAUNCHES E-VISA FOR CHINESE TOURISTS Minister of Tourism & Culture Malaysia Dato’ Seri Mohamed Nazri Abdul Aziz (middle) flanked by the Ambassador of Malaysia to the People's Republic of China Dato' Zainuddin Yahya (left) and the Deputy Undersecretary of the Ministry of Home Affairs Malaysia Azman Azra Abdul Rahman at the launching ceremony. BEIJING, 1 March 2016: The Minister of Tourism and Culture Malaysia Dato’ Seri Mohamed Nazri Abdul Aziz officially launched Malaysia’s e-Visa programme today. The visa exemption and e-Visa programme is introduced by the Malaysian government to facilitate tourists from China travelling to Malaysia. Implementation of the e-Visa system is the key initiative to help accomplish the goal of further expanding and developing the tourism industries of both Malaysia and China. The implementation of the e-Visa system will be carried out in three phases starting with Chinese nationals residing in Mainland China, followed by the second phase that includes Chinese nationals residing outside Mainland China, and eventually with the final phase, which incorporates other countries such as India, Myanmar, Nepal and Sri Lanka. MALAYSIA TOURISM PROMOTION BOARD (MINISTRY OF TOURISM & CULTURE, MALAYSIA) No. 2, Tower 1, Jalan P5/6, Precinct 5, 62200 Putrajaya, Malaysia Tel: +603 8891 8000; Official: malaysia.travel; Corporate: tourism.gov.my Facebook: friendofmalaysia; Twitter: tourismmalaysia; Blog: blog.tourism.gov.my 1 ! The e-Visa is for the purpose of travelling and visiting relatives and friends only, with the length of stay limited to 30 days and for a single entry to Malaysia within three months from the date of issuance of the e-Visa. -

Press Release
PRESS RELEASE United Nations Secretary-General appoints the Japanese singer MISIA as Honorary Ambassador for the 2010 United Nations Biodiversity Conference Montreal, 1 March 2010. United Nations Secretary-General Ban Ki-moon has appointed the Japanese female singer and songwriter, MISIA, as the Honorary Ambassador for the forthcoming tenth meeting of the Conference of the Parties to the Convention on Biological Diversity, which will be held from 18 to 29 October 2010 in Nagoya, Aichi Prefecture, Japan. In her honorary role, MISIA will help raise awareness of the continuing depletion of biological resources and inform the public on the sustainable use of biodiversity. “In this International Year of Biodiversity, we must counter the perception that people are disconnected from our natural environment. We must increase understanding of the implications of losing biodiversity,” said Secretary-General Ban Ki-moon. Referring to Honorary Ambassador MISIA, the Secretary-General said she could “help highlight our common struggles for protecting life on Earth and the actions we can take to slow down biodiversity loss.” Throughout her successful career, MISIA has been an advocate of sustainable development. Through her performances, she has worked to raise public awareness on the United Nations Millennium Development Goals and, in 2008, she established a voluntary organization, Child AFRICA, to support education in Africa. This honorary appointment will help bring much needed attention to the importance of biodiversity and the urgent need for progress to meet the three objectives of the Convention on Biological Diversity (CBD). On learning of her appointment, MISIA said, “While travelling to different parts of the world, I have realized that our life and culture vary a lot and this diversity has been nurtured by the rich natural environments of different regions. -

2020 China Country Profile
1 China Music Industry Development Report COUNTRY China: statistics PROFILE MARKET PROFILE claiming that China’s digital music business 13.08.20 ❱China increased by 5.5% from 2017 to 2018 while the number of digital music users exceeded Population... 1.4bn 550m, a jump of 5.1% year-on-year. GDP (purchasing power parity)... $25.36tn GDP per capita (PPP)... $18,200 What, exactly, these digital music users are doing is still not entirely clear. Streaming, as 904m Internet users... you might imagine, dominates digital music Broadband connections... 407.39m consumption in China. The divide, however, Broadband - subscriptions per 100 inhabitants... 29 between ad-supported and paid users is opaque at best. The IFPI, for example, reports Mobile phone subscriptions... 1.65bn that revenue from ad-supported streaming Smartphone users... 781.7m is greater than that of subscription in China Sources: CIA World Factbook/South China Morning Post/Statista – a claim that was met with some surprise by some local Music Ally sources. “For most of the platforms, subscription income is still the largest contributor in the digital music market,” says a representative of NetEase A lack of reliable figures makes China a Cloud Music. difficult music market to understand. Even so, Simon Robson, president, Asia Region, its digital potential is vast. Warner Music, says that the ad-supported China basically lives model isn’t really set up to make money in China but is instead designed to drive traffic CHINA’S GROWING IMPORTANCE as a service NetEase Cloud Music, warned inside WeChat now... to the service. “Within China, the GDP is so digital music market is perhaps only matched that IFPI numbers for China were largely we fix problems for the different from city to city that it becomes by a fundamental lack of understanding based on advances; that means they may difficult to set standard pricing per month that exists about the country. -

Press Release 20 November 2018 Winners of Music Moves Europe Talent Awards Revealed International Jury Selects Twelve Winners T
Press release 20 November 2018 Winners of Music Moves Europe Talent Awards revealed International jury selects twelve winners • Public choice voting opens Today The first winners of the Music Moves Europe Talent Awards have been revealed. Following the announcement of the twenty-four nominees at Reeperbahn, an international jury selected twelve winners, who represent the European sound of today and tomorrow. The winners are: Bishop Briggs (UK) and Lxandra (FI) [Pop], Pale Waves (UK) and Pip Blom (NL) [Rock], Smerz (NO) Stelartronic (AT) [Electronic], Rosalía (ES) and Aya Nakamura (FR) [RnB/Urban], Blackwave. (BE), Reykjavíkurdætur (IS) [Hip Hop/Rap], Avec (AT) and Albin Lee Meldau (SE) [Singer-songwriter]. The Music Moves Europe Talent Awards are the new European Union Prize for popular and contemporary music. The prizes will be awarded at ESNS (Eurosonic Noorderslag) in Groningen. Additionally, the general public will select another winner from each category through the Public Choice Award through online voting, starting today. The Public Choice Award winners will be revealed at the award ceremony on Wednesday January 16, 2019. The Jury The international jury for the Music Moves Europe Talent Awards consists of Huw Stephens (Presenter BBC Radio 1), Julia Gudzent (Festival Programmer Melt!), Katia Giampaolo (CEO Estragon Club Italy), Kristian Kostov (Winner EBBA + Public Choice Award 2018) and Wilbert Mutsaers (Head of Content, Spotify Benelux). Representing the organizing parties are Robert Meijerink (Booker ESNS) as the chairman of the Jury and Bjørn Pfarr (Booker Reeperbahn Festival) as the vice-chairman of the jury. The Prize The aim of the Music Moves Europe Talent Awards is to celebrate new and upcoming artists from Europe and support them in order to help them develop and accelerate their international careers. -

Chinese Music Reality Shows: a Case Study
Chinese Music Reality Shows: A Case Study A Thesis Submitted to the Faculty of Drexel University by Zhengyuan Bi In partial fulfillment of the requirements for the degree of Master of Science in Television Management January 2017 ii © Copyright 2017 Zhengyuan Bi All Right Reserved. iii DEDICATION I dedicate this thesis to my family and my friends, with a special feeling of gratitude to my loving parents, my friends Queena Ai, Eileen Zhou and Lili Mao, and my former boss Kenny Lam. I will always appreciate their love and support! iv ACKNOWLEDGMENTS I would like to take this opportunity to thank my thesis advisor Philip Salas and Television Management Program Director Al Tedesco for their great support and guidance during my studies at Drexel University. I would also like to thank Katherine Houseman for her kind support and assistance. I truly appreciate their generous contribution to me and all the students. I would also like to thank all the faculty of Westphal College of Media Arts & Design, all the classmates that have studied with me for these two years. We are forever friends and the best wishes to each of you! v Table of Contents DEDICATION ………………………………………………………………………………..iii ACKNOWLEDGEMENTS…………………………………………………………………...iv ABSTRACT……………………………………………………………………………………vii Chapter 1: Introduction………………………………………………………………………..1 1.1 Introduction………………………………………………………………………………...3 1.2 Statement of the Problem…………………………………………….................................3 1.3 Background…………………………………………………………………………………4 1.4 Purpose of the study…………………………………………………………………….....5 -

Eurovision 2015
Eurovision 2015 The Eurovision Song Contest is an annual event organized by the European Broadcasting Union. It brings together the members of the Union as part of a musical competition, broadcast live and simultaneously by all participating broadcasters. The first edition of the competition took place on 24th May 1956 in Lugano, Switzerland. Then seven founding countries competed for victory. Since then, the competition was held every year without interruption. This year’s we are the 60 edition of this concours. At present, the competition remains one of the oldest TV programs in the world and the largest ever organized musical contest. Its success has gone beyond the borders of the European continent and it has inspired many other musical competitions. Participating countries Did not qualify from the semi final Countries that participated in the past but not in 2015 Map of participating countries The official list of participants was unveiled 23th December 2014. This list shows the participation of thirty-nine countries for this 60th edition. Also we can see that three countries have had their new appearances: Serbia Cyprus The Czech Republic The participation of Greece was subject to much debate. In 2013, Greece no longer had any broadcaster EBU member and an exemption for participation in the Eurovision could not be reallocated to the country. It was not until the General Assembly of the EBU, on 4th and 5th December 2014 for a decision on whether or not the Greek group becomes a member of the European institution. Finally, the candidature has accepted, their participation is possible and so that in the coming years. -

Parent-Singer Handbook 2019 – 2020 (Children’S Chorus, Treble Choir, Chorale & Concert Choirs)
Parent-Singer Handbook 2019 – 2020 (Children’s Chorus, Treble Choir, Chorale & Concert Choirs) Honors Choirs of Southeast Minnesota Assisi Heights, Suite 920 1001 14th Street NW Rochester, MN 55901 Office Phone: (507) 252-0505 Office E-mail: [email protected] Web Site: www.HonorsChoirs.org Office Hours Monday-Thursday: 10:00am - 4:00pm (closed Fridays) Updated: 5/22/19 HONORS CHOIRS ORGANIZATION Mission Statement The mission of the Honors Choirs of Southeast Minnesota is to promote the highest standard of excellence in the preparation and performance of choral music, seeking to provide artistic challenge and growth opportunities for youth throughout the region and enjoyment for the community at large. Conductors & Music Staff Rick Kvam, Artistic Director Rick Kvam, conductor-Concert Choir men & women, gr. 10 – 12 [email protected] Aaron Schumacher, conductor-Chorale boys & girls, gr. 7 – 9 [email protected] Bill Nelson, conductor-Treble Choir boys & girls, gr. 5-6 [email protected] Amy Nelson, conductor-Children’s Chorus boys & girls, gr. 3-5 [email protected] Korrie Johnson, instructor-Half Notes boys & girls, gr. 1-2 [email protected] Jan Kvam, pianist Concert Choir & Treble Choir Jon Davis, pianist Chorale & Half Notes Cindy Breederland, pianist Children’s Chorus Administrative Staff Jayne Rothschild Executive Director [email protected] Mary Wille Lead Administrator [email protected] Karri Campbell Finance Administrator [email protected] 2019-20 Board of Directors Melissa Saunders– President Bruce Bonnicksen Dotti Loutfi Anjanette Bandel – Vice President Amy Crockett Jeff Pieters Anna Sanchez – Secretary Jolene Hansen Valerie Presa Chuck Johnson – Treasurer Amelia Harthan Chris Rowen Heidi Dieter– Past-President Rafael Jimenez Dan Tschumperlin Bradley Nuss Mary Vogel Janine Yanisch The Board of Directors generally meets every third Monday of each month except December and July. -

Dimash Kudaibergen Becomes 1St Kazakh Artist to Participate in SCI Presidential Inauguration Celebrations in the U.S
FOR IMMEDIATE RELEASE January 6, 2021 MEDIA CONTACT Jina Shim [email protected] 202-347-4876 WASHINGTON, DC (January 6, 2021) Dimash Kudaibergen Becomes 1st Kazakh Artist to Participate in SCI Presidential Inauguration Celebrations in the U.S. Sister Cities International (SCI) is proud to announce that Dimash Kudaibergen will be performing at the SCI Presidential Inaugural Celebration on January 17th, 2021. “We are beyond thrilled and excited that Dimash will be joining our SCI family to formally recognize President-Elect Joe Biden and the incoming administration,” said Leroy Allala, President and CEO of Sister Cities International. “His participation in our event further strengthens the ties between Kazakhstan and the United States.” This will be the first time in SCI’s 67-year history and since U.S./KZ relations began (almost 30 years ago) that an artist from Kazakhstan has collaborated with a U.S. organization for such an occasion. Sister Cities International’s Inaugural event will include the participation of the diplomatic corps and launches our week of presidential inaugural celebrations. Due to the current pandemic, this year’s event will be held virtually on Sunday, January 17th from 7:00 PM to 9:00 PM EST. Since SCI’s founding in 1956 by President Dwight Eisenhower, each president has been recognized as its honorary co-chair. With this coming inauguration, President-Elect Joe Biden will be named as the honorary chairman for Sister Cities International. Dimash Kudaibergen is a Kazakh singer and songwriter who is university trained in classical, as well as contemporary music. Truly an international artist, he has performed songs in twelve different languages.