Computational Study of Transcriptional Regulation - from Sequence to Expression
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Applied Category Theory for Genomics – an Initiative
Applied Category Theory for Genomics { An Initiative Yanying Wu1,2 1Centre for Neural Circuits and Behaviour, University of Oxford, UK 2Department of Physiology, Anatomy and Genetics, University of Oxford, UK 06 Sept, 2020 Abstract The ultimate secret of all lives on earth is hidden in their genomes { a totality of DNA sequences. We currently know the whole genome sequence of many organisms, while our understanding of the genome architecture on a systematic level remains rudimentary. Applied category theory opens a promising way to integrate the humongous amount of heterogeneous informations in genomics, to advance our knowledge regarding genome organization, and to provide us with a deep and holistic view of our own genomes. In this work we explain why applied category theory carries such a hope, and we move on to show how it could actually do so, albeit in baby steps. The manuscript intends to be readable to both mathematicians and biologists, therefore no prior knowledge is required from either side. arXiv:2009.02822v1 [q-bio.GN] 6 Sep 2020 1 Introduction DNA, the genetic material of all living beings on this planet, holds the secret of life. The complete set of DNA sequences in an organism constitutes its genome { the blueprint and instruction manual of that organism, be it a human or fly [1]. Therefore, genomics, which studies the contents and meaning of genomes, has been standing in the central stage of scientific research since its birth. The twentieth century witnessed three milestones of genomics research [1]. It began with the discovery of Mendel's laws of inheritance [2], sparked a climax in the middle with the reveal of DNA double helix structure [3], and ended with the accomplishment of a first draft of complete human genome sequences [4]. -

Functional Effects Detailed Research Plan
GeCIP Detailed Research Plan Form Background The Genomics England Clinical Interpretation Partnership (GeCIP) brings together researchers, clinicians and trainees from both academia and the NHS to analyse, refine and make new discoveries from the data from the 100,000 Genomes Project. The aims of the partnerships are: 1. To optimise: • clinical data and sample collection • clinical reporting • data validation and interpretation. 2. To improve understanding of the implications of genomic findings and improve the accuracy and reliability of information fed back to patients. To add to knowledge of the genetic basis of disease. 3. To provide a sustainable thriving training environment. The initial wave of GeCIP domains was announced in June 2015 following a first round of applications in January 2015. On the 18th June 2015 we invited the inaugurated GeCIP domains to develop more detailed research plans working closely with Genomics England. These will be used to ensure that the plans are complimentary and add real value across the GeCIP portfolio and address the aims and objectives of the 100,000 Genomes Project. They will be shared with the MRC, Wellcome Trust, NIHR and Cancer Research UK as existing members of the GeCIP Board to give advance warning and manage funding requests to maximise the funds available to each domain. However, formal applications will then be required to be submitted to individual funders. They will allow Genomics England to plan shared core analyses and the required research and computing infrastructure to support the proposed research. They will also form the basis of assessment by the Project’s Access Review Committee, to permit access to data. -

Hmms Representing All Proteins of Known Structure. SCOP Sequence Searches, Alignments and Genome Assignments Julian Gough* and Cyrus Chothia
268–272 Nucleic Acids Research, 2002, Vol. 30, No. 1 © 2002 Oxford University Press SUPERFAMILY: HMMs representing all proteins of known structure. SCOP sequence searches, alignments and genome assignments Julian Gough* and Cyrus Chothia MRC Laboratory of Molecular Biology, Hills Road, Cambridge CB2 2QH, UK Received September 28, 2001; Revised and Accepted October 30, 2001 ABSTRACT (currently 59) covering approximately half of the The SUPERFAMILY database contains a library of soluble protein domains. The assignments, super- family breakdown and statistics on them are available hidden Markov models representing all proteins of from the server. The database is currently used by known structure. The database is based on the SCOP this group and others for genome annotation, structural ‘superfamily’ level of protein domain classification genomics, gene prediction and domain-based genomic which groups together the most distantly related studies. proteins which have a common evolutionary ancestor. There is a public server at http://supfam.org which provides three services: sequence searching, INTRODUCTION multiple alignments to sequences of known structure, The SUPERFAMILY database is based on the SCOP (1) and structural assignments to all complete genomes. classification of protein domains. SCOP is a structural domain- Given an amino acid or nucleotide query sequence based heirarchical classification with several levels including the server will return the domain architecture and the ‘superfamily’ level. Proteins grouped together at the super- SCOP classification. The server produces alignments family level are defined as having structural, functional and sequence evidence for a common evolutionary ancestor. It is at of the query sequences with sequences of known this level, as the name suggests, that SUPERFAMILY operates structure, and includes multiple alignments of because it is the level with the most distantly related protein genome and PDB sequences. -

Ontology-Based Methods for Analyzing Life Science Data
Habilitation a` Diriger des Recherches pr´esent´ee par Olivier Dameron Ontology-based methods for analyzing life science data Soutenue publiquement le 11 janvier 2016 devant le jury compos´ede Anita Burgun Professeur, Universit´eRen´eDescartes Paris Examinatrice Marie-Dominique Devignes Charg´eede recherches CNRS, LORIA Nancy Examinatrice Michel Dumontier Associate professor, Stanford University USA Rapporteur Christine Froidevaux Professeur, Universit´eParis Sud Rapporteure Fabien Gandon Directeur de recherches, Inria Sophia-Antipolis Rapporteur Anne Siegel Directrice de recherches CNRS, IRISA Rennes Examinatrice Alexandre Termier Professeur, Universit´ede Rennes 1 Examinateur 2 Contents 1 Introduction 9 1.1 Context ......................................... 10 1.2 Challenges . 11 1.3 Summary of the contributions . 14 1.4 Organization of the manuscript . 18 2 Reasoning based on hierarchies 21 2.1 Principle......................................... 21 2.1.1 RDF for describing data . 21 2.1.2 RDFS for describing types . 24 2.1.3 RDFS entailments . 26 2.1.4 Typical uses of RDFS entailments in life science . 26 2.1.5 Synthesis . 30 2.2 Case study: integrating diseases and pathways . 31 2.2.1 Context . 31 2.2.2 Objective . 32 2.2.3 Linking pathways and diseases using GO, KO and SNOMED-CT . 32 2.2.4 Querying associated diseases and pathways . 33 2.3 Methodology: Web services composition . 39 2.3.1 Context . 39 2.3.2 Objective . 40 2.3.3 Semantic compatibility of services parameters . 40 2.3.4 Algorithm for pairing services parameters . 40 2.4 Application: ontology-based query expansion with GO2PUB . 43 2.4.1 Context . 43 2.4.2 Objective . -

AUTOPHY by Deepika Prasad a Thesis
ANALYZING MARKER GENE DIVERSITY USING AN AUTOMATED PHYLOGENETIC TOOL: AUTOPHY by Deepika Prasad A thesis submitted to the Faculty of the University of Delaware in partial fulfillment of the requirements for the degree of Master of Science in Bioinformatics and Computational Biology Spring 2017 © 2017 Deepika Prasad All Rights Reserved ANALYZING MARKER GENE DIVERSITY USING AN AUTOMATED PHYLOGENETIC TOOL: AUTOPHY by Deepika Prasad Approved: __________________________________________________________ Shawn Polson, Ph.D. Professor in charge of thesis on behalf of the Advisory Committee Approved: __________________________________________________________ Kathleen F.McCoy, Ph.D. Chair of the Department of Computer and Information Sciences Approved: __________________________________________________________ Babatunde A. Ogunnaike, Ph.D. Dean of the College of Engineering Approved: __________________________________________________________ Ann L. Ardis, Ph.D. Senior Vice Provost for Graduate and Professional Education ACKNOWLEDGMENTS I want to express my sincerest gratitude to Professor Shawn Polson for his patience, continuous support, and enthusiasm. I could not have imagined having a better mentor to guide me through the process of my Master’s thesis. I would like to thank my committee members, Professor Eric Wommack and Honzhan Huang for their insightful comments and guidance in the thesis. I would also like to express my gratitude to Barbra Ferrell for asking important questions throughout my thesis research and helping me with the writing process. I would like to thank my friend Sagar Doshi, and lab mates Daniel Nasko, and Prasanna Joglekar, for helping me out with data, presentations, and giving important advice, whenever necessary. Last but not the least, I would like to thank my family, for being my pillars of strength. -

PREDICTD: Parallel Epigenomics Data Imputation with Cloud-Based Tensor Decomposition
bioRxiv preprint doi: https://doi.org/10.1101/123927; this version posted April 4, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. PREDICTD: PaRallel Epigenomics Data Imputation with Cloud-based Tensor Decomposition Timothy J. Durham Maxwell W. Libbrecht Department of Genome Sciences Department of Genome Sciences University of Washington University of Washington J. Jeffry Howbert Jeff Bilmes Department of Genome Sciences Department of Electrical Engineering University of Washington University of Washington William Stafford Noble Department of Genome Sciences Department of Computer Science and Engineering University of Washington April 4, 2017 Abstract The Encyclopedia of DNA Elements (ENCODE) and the Roadmap Epigenomics Project have produced thousands of data sets mapping the epigenome in hundreds of cell types. How- ever, the number of cell types remains too great to comprehensively map given current time and financial constraints. We present a method, PaRallel Epigenomics Data Imputation with Cloud-based Tensor Decomposition (PREDICTD), to address this issue by computationally im- puting missing experiments in collections of epigenomics experiments. PREDICTD leverages an intuitive and natural model called \tensor decomposition" to impute many experiments si- multaneously. Compared with the current state-of-the-art method, ChromImpute, PREDICTD produces lower overall mean squared error, and combining methods yields further improvement. We show that PREDICTD data can be used to investigate enhancer biology at non-coding human accelerated regions. PREDICTD provides reference imputed data sets and open-source software for investigating new cell types, and demonstrates the utility of tensor decomposition and cloud computing, two technologies increasingly applicable in bioinformatics. -

Advancing a Systems Cell-Free Metabolic Engineering Approach to Natural Product Synthesis and Discovery
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 12-2020 Advancing a systems cell-free metabolic engineering approach to natural product synthesis and discovery Benjamin Mohr University of Tennessee Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Recommended Citation Mohr, Benjamin, "Advancing a systems cell-free metabolic engineering approach to natural product synthesis and discovery. " PhD diss., University of Tennessee, 2020. https://trace.tennessee.edu/utk_graddiss/6837 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Benjamin Mohr entitled "Advancing a systems cell-free metabolic engineering approach to natural product synthesis and discovery." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Doctor of Philosophy, with a major in Energy Science and Engineering. Mitchel Doktycz, Major Professor We have read this dissertation and recommend its acceptance: Jennifer Morrell-Falvey, Dale Pelletier, Michael Simpson, Robert Hettich Accepted for the Council: Dixie L. Thompson Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) Advancing a systems cell-free metabolic engineering approach to natural product synthesis and discovery A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville Benjamin Pintz Mohr December 2019 c by Benjamin Pintz Mohr, 2019 All Rights Reserved. -

ACCEPTED PAPERS – ABSTRACTS (As of May 5, 2006) Comparative
ACCEPTED PAPERS – ABSTRACTS (as of May 5, 2006) Comparative Genomics Hairpins in a haystack: recognizing microrna precursors in comparative genomics data Author(s): Jana Hertel, Peter F. Stadler Recently, genome wide surveys for non-coding RNAs have provided evidence for tens of thousands of previously undescribed evolutionary conserved RNAs with distinctive secondary structures. The annotation of these putative ncRNAs, however, remains a difficult problem. Here we describe an SVM-based approach that, in conjunction with a non-stringent filter for consensus secondary structures, is capable of efficiently recognizing microRNA precursors in multiple sequence alignments. The software was applied to recent genome-wide RNAz surveys of mammals, urochordates, and nematodes. Keywords: miRNA, support vector machine, non-coding RNA Comparative Genomics Comparative genomics reveals unusually long motifs in mammalian genomes Author(s): Neil Jones, Pavel Pevzner Motivation: The recent discovery of the first small modulatory RNA (smRNA) presents the challenge of finding other molecules of similar length and conservation level. Unlike short interfering RNA (siRNA) and micro-RNA (miRNA), effective computational and experimental screening methods are not currently known for this species of RNA molecule, and the discovery of the one known example was partly fortuitous because it happened to be complementary to a well-studied DNA binding motif (the Neuron Restrictive Silencer Element). Results: The existing comparative genomics approaches (e.g., phylogenetic footprinting) rely on alignments of orthologous regions across multiple genomes. This approach, while extremely valuable, is not suitable for finding motifs with highly diverged ``non-alignable'' flanking regions. Here we show that several unusually long and well conserved motifs can be discovered de novo through a comparative genomics approach that does not require an alignment of orthologous upstream regions. -

Daniel Aalberts Scott Aa
PLOS Computational Biology would like to thank all those who reviewed on behalf of the journal in 2015: Daniel Aalberts Jeff Alstott Benjamin Audit Scott Aaronson Christian Althaus Charles Auffray Henry Abarbanel Benjamin Althouse Jean-Christophe Augustin James Abbas Russ Altman Robert Austin Craig Abbey Eduardo Altmann Bruno Averbeck Hermann Aberle Philipp Altrock Ferhat Ay Robert Abramovitch Vikram Alva Nihat Ay Josep Abril Francisco Alvarez-Leefmans Francisco Azuaje Luigi Acerbi Rommie Amaro Marc Baaden Orlando Acevedo Ettore Ambrosini M. Madan Babu Christoph Adami Bagrat Amirikian Mohan Babu Frederick Adler Uri Amit Marco Bacci Boris Adryan Alexander Anderson Stephen Baccus Tinri Aegerter-Wilmsen Noemi Andor Omar Bagasra Vera Afreixo Isabelle Andre Marc Baguelin Ashutosh Agarwal R. David Andrew Timothy Bailey Ira Agrawal Steven Andrews Wyeth Bair Jacobo Aguirre Ioan Andricioaei Chris Bakal Alaa Ahmed Ioannis Androulakis Joseph Bak-Coleman Hasan Ahmed Iris Antes Adam Baker Natalie Ahn Maciek Antoniewicz Douglas Bakkum Thomas Akam Haroon Anwar Gabor Balazsi Ilya Akberdin Stefano Anzellotti Nilesh Banavali Eyal Akiva Miguel Aon Rahul Banerjee Sahar Akram Lucy Aplin Edward Banigan Tomas Alarcon Kevin Aquino Martin Banks Larissa Albantakis Leonardo Arbiza Mukul Bansal Reka Albert Murat Arcak Shweta Bansal Martí Aldea Gil Ariel Wolfgang Banzhaf Bree Aldridge Nimalan Arinaminpathy Lei Bao Helen Alexander Jeffrey Arle Gyorgy Barabas Alexander Alexeev Alain Arneodo Omri Barak Leonidas Alexopoulos Markus Arnoldini Matteo Barberis Emil Alexov -

Computational Methods for Cis-Regulatory Module Discovery A
Computational Methods for Cis-Regulatory Module Discovery A thesis presented to the faculty of the Russ College of Engineering and Technology of Ohio University In partial fulfillment of the requirements for the degree Master of Science Xiaoyu Liang November 2010 © 2010 Xiaoyu Liang. All Rights Reserved. 2 This thesis titled Computational Methods for Cis-Regulatory Module Discovery by XIAOYU LIANG has been approved for the School of Electrical Engineering and Computer Science and the Russ College of Engineering and Technology by Lonnie R.Welch Professor of Electrical Engineering and Computer Science Dennis Irwin Dean, Russ College of Engineering and Technology 3 ABSTRACT LIANG, XIAOYU, M.S., November 2010, Computer Science Computational Methods for Cis-regulatory Module Discovery Director of Thesis: Lonnie R.Welch In a gene regulation network, the action of a transcription factor binding a short region in non-coding sequence is reported and believed as the key that triggers, or represses genes’ expression. Further analysis revealed that, in higher organisms, multiple transcription factors work together and bind multiple sites that are located nearby in genomic sequences, rather than working alone and binding a single anchor. These multiple binding sites in the non-coding region are called cis -regulatory modules. Identifying these cis - regulatory modules is important for modeling gene regulation network. In this thesis, two methods have been proposed for addressing the problem, and a widely accepted evaluation was applied for assessing the performance. Additionally, two practical case studies were completed and reported as the application of the proposed methods. Approved: _____________________________________________________________ Lonnie R.Welch Professor of Electrical Engineering and Computer Science 4 ACKNOWLEDGEMENTS I would like to express my sincere gratitude to my advisor, Dr. -
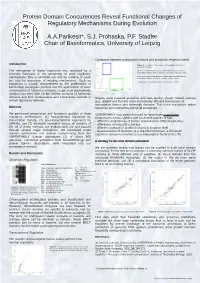
Protein Domain Coocurences Reveal Functional Changes of Regulatory Mechanisms During Evolution
Protein Domain Coocurences Reveal Functional Changes of Regulatory Mechanisms During Evolution A.A.Parikesit*, S.J. Prohaska, P.F. Stadler Chair of Bioinformatics, University of Leipzig Correlation between transcription factors and chromatin related proteins Introduction Figure 3 : Correlation of the number of transcription factors and chromatin domains. The emergence of higher organisms was facilitated by a Blue box. Species with few chromatin-related domains but many dramatic increases in the complexity of gene regulatory transcription factors: human, kangaroo rat, mouse, opossum, fugu. Green box: species with many chromatin-related domains but few mechanisms. This is achieved not only by addition of novel transcription factors: seq squirt, medaka, dolphin, yeast but also by expansion of existing mechanisms. Such an There are no organisms (known) that have a lot of both. expansion is usually characterized by the proliferation of functionally paralogous proteins and the appearance of novel combinations of functional domains. Large scale phylogenetic analysis can shed light on the relative amounts of functional domains and their combinations and interactions involved in Results show massive problems with data quality: closely related species certain regulatory networks. (e.g, dolphin and human) show dramatically different distributions of transcription factors and chromatin domains. This is not reasonable within Methods mammals and contradicts biological knowledge. We performed comparative and functional analysis of three SUPERFAMILY thus cannot be used for large-scale quantitative regulatory mechanisms: (1) transcriptional regulation by comparisons across species due to several sources of bias: transcription factors, (2) post-transcriptional regulation by - different completeness of protein annotation for different genomes miRNAs, and (3) chromatin regulation across all domains of - differences in transcript coverage life. -

Motif Selection Using Simulated Annealing Algorithm with Application to Identify Regulatory Elements
Motif Selection Using Simulated Annealing Algorithm with Application to Identify Regulatory Elements A thesis presented to the faculty of the Russ College of Engineering and Technology of Ohio University In partial fulfillment of the requirements for the degree Master of Science Liang Chen August 2018 © 2018 Liang Chen. All Rights Reserved. 2 This thesis titled Motif Selection Using Simulated Annealing Algorithm with Application to Identify Regulatory Elements by LIANG CHEN has been approved for the Department of Electrical Engineering and Computer Science and the Russ College of Engineering and Technology by Lonnie Welch Professor of Electrical Engineering and Computer Science Dennis Irwin Dean, Russ College of Engineering and Technology 3 Abstract CHEN, LIANG, M.S., August 2018, Computer Science Master Program Motif Selection Using Simulated Annealing Algorithm with Application to Identify Regulatory Elements (106 pp.) Director of Thesis: Lonnie Welch Modern research on gene regulation and disorder-related pathways utilize the tools such as microarray and RNA-Seq to analyze the changes in the expression levels of large sets of genes. In silico motif discovery was performed based on the gene expression profile data, which generated a large set of candidate motifs (usually hundreds or thousands of motifs). How to pick a set of biologically meaningful motifs from the candidate motif set is a challenging biological and computational problem. As a computational problem it can be modeled as motif selection problem (MSP). Building solutions for motif selection problem will give biologists direct help in finding transcription factors (TF) that are strongly related to specific pathways and gaining insights of the relationships between genes.