Chemical and Metabolomic Analyses of Cuprizone-Induced
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download Download
J. Pharm. Tech. Res. Management Vol. 8, No. 1 (2020), pp.39–46 Vol. 7 | No. 2 | Nov 2019 Journal of Pharmaceutical Technology Research and Management Journal homepage: https://jptrm.chitkara.edu.in/ Ranitidine Induced Hepatotoxicity: A Review Amit Bandyopadhyay Banerjee1, Manisha Gupta2, Thakur Gurjeet Singh3, Sandeep Arora4 and Onkar Bedi5* Chitkara College of Pharmacy, Chitkara University, Punjab-140401, India [email protected] [email protected] [email protected] [email protected] 5*[email protected] (Corresponding Author) ARTICLE INFORMATION ABSTRACT Received: January 29, 2020 Background: Ranitidine (RAN) is one of the common drugs associated with idiosyncratic Revised: April 08, 2020 adverse drug reactions (IADRs) in humans. It was found to be associated with severe adverse drug Accepted: April 28, 2020 reactions due to the presence of contaminants such as N-Nitrosodimethylamine (NDMA) which Published Online: May 20, 2020 is claimed to be carcinogenic. As a consequence, on April 1, 2020, United States Food and Drug Keywords: Administration (USFDA) had decided to call off all the RAN products from the market. The exact DILI, Ranitidine withdrawal, RAN induced cause of RAN associated idiosyncratic hepatotoxicity is not clear yet. hepatotoxicity Purpose: To summarize and analyze the reason behind the withdrawal of RAN products from the market and whether ranitidine will be available again in future or will FDA withdraw approvals of ranitidine National Drug Authority (NDA) and an abbreviated new drug application (ANDA)? Methods: We performed a systematic PubMed/MEDLINE search of studies investigating the reason behind the withdrawal of RAN products and explored the possible mechanism associated with RAN induced hepatotoxicity. -

BUREAU CIRCULAR No. 12 S. 1997
REPUBLIC OF THE PHILIPPINES DEPARTMENT OF HEALTH BUREAU OF FOOD AND DRUGS D.O.H. Compound Alabang, Muntinlupa Metro Manila September 4, 1997 BUREAU CIRCULAR No. 12 series 1997 TO : ALL COSMETIC MANUFACTURERS, TRADERS, IMPORTERS AND PARTIES CONCERNED SUBJECT : 1997 UPDATED LISTING OF COSMETIC INGREDIENTS The BFAD Management Committee, in its meeting on August 28, 1997 has adopted and approved the updated technical standards and requirements set for cosmetic ingredients as recommended by the Joint BFAD and Cosmetic Industry Study Group’s Technical Committee. As such, the sections of Bureau Circular No. 19-A series of 1997 hereunder described are amended by the listings of cosmetic ingredients hereunto appended, to wit: Section III - Restricted Ingredients For Use In Cosmetics Table I : List Of Substances Which Cosmetics Products Must Not Contain Except Subject To The Restrictions And Conditions Specified Table II : List Of Preservatives Which Cosmetic Products May Contain Subject To The Restrictions And Conditions Specified Table III : List Of Preservatives Provisionally Allowed Section IV - Non-Permissible Colors In Cosmetics Table IV : Non-Permissible Colors In Cosmetics Section V - Permissible Color Additives Table V : Permissible Color Additives; List Of Colorants Restricted/Allowed For Cosmetic Products Table VI : List Of Provisionally Allowed Colors In Cosmetic Preparations Section VI: Sunscreen Agents Table VII : List Of Sunscreen Agents Which Cosmetics Products May Contain Table VIII : List Of Sunscreen Agents Which Cosmetics Products May Provisionally Contain Table IX : List Of Substances Which Must Not Form Part Of The Composition Of Cosmetic Products Additionally, BFAD has decided to extend the approval of products containing benzethonium chloride only up to December 31, 1997 in view of the ban of the said ingredient under the European Economic Community’s (EEC) August 1996 Directive, unless additional safety data on the same are found before December 31, 1997. -

Pdf; Chi 2015 DPP Air in Cars.Pdf; Dodson 2014 DPP Dust CA.Pdf; Kasper-Sonnenberg 2014 Phth Metabolites.Pdf; EU Cosmetics Regs 2009.Pdf
Bouge, Cathy (ECY) From: Nancy Uding <[email protected]> Sent: Friday, January 13, 2017 10:24 AM To: Steward, Kara (ECY) Cc: Erika Schreder Subject: Comments re. 2016 CSPA Rule Update - DPP Attachments: DPP 131-18-0 exposure.pdf; Chi 2015 DPP air in cars.pdf; Dodson 2014 DPP dust CA.pdf; Kasper-Sonnenberg 2014 phth metabolites.pdf; EU Cosmetics Regs 2009.pdf Please accept these comments from Toxic-Free Future concerning the exposure potential of DPP for consideration during the 2016 CSPA Rule update. Regards, Nancy Uding -- Nancy Uding Grants & Research Specialist Toxic-Free Future 206-632-1545 ext.123 http://toxicfreefuture.org 1 JES-00888; No of Pages 9 JOURNAL OF ENVIRONMENTAL SCIENCES XX (2016) XXX– XXX Available online at www.sciencedirect.com ScienceDirect www.elsevier.com/locate/jes Determination of 15 phthalate esters in air by gas-phase and particle-phase simultaneous sampling Chenchen Chi1, Meng Xia1, Chen Zhou1, Xueqing Wang1,2, Mili Weng1,3, Xueyou Shen1,4,⁎ 1. College of Environmental & Resource Sciences, Zhejiang University, Hangzhou 310058, China 2. Zhejiang National Radiation Environmental Technology Co., Ltd., Hangzhou 310011, China 3. School of Environmental and Resource Sciences, Zhejiang Agriculture and Forestry University, Hangzhou 310058, China 4. Zhejiang Provincial Key Laboratory of Organic Pollution Process and Control, Hangzhou 310058, China ARTICLE INFO ABSTRACT Article history: Based on previous research, the sampling and analysis methods for phthalate esters (PAEs) Received 24 December 2015 were improved by increasing the sampling flow of indoor air from 1 to 4 L/min, shortening the Revised 14 January 2016 sampling duration from 8 to 2 hr. -

Col.V.MISSION of the EUROPEAN COMMUNITIES
COl.V.MISSION OF THE EUROPEAN COMMUNITIES SEC(90) 1985 final· Brussels, 29 October 1990 Proposa I for a COUNCIL DIRECTIVE on the approximation of the laws of the Member States relat.ing to cosmetic products -2- EXPLANATORY UEUORANDUM 1. In the context of a people's Europe, the Commission attaches great Importance to simplifying and clarifying Community law so as to make It clearer and more accessible to the ordinary citizen, thus giving hIm new opportunItIes and the chance to make use of the spec if i c rights It gives him. This aim cannot be achieved so long as numerous provisions that have been amended several times, often QUite substantially, remain scattered, so that they must be sought partly In the original Instrument and partly In later amending ones. Considerable research work, comparing many different Instruments, Is thus needed to Identify the current rules. For this reason a consot tdatton of rules that have freQuently been amended Is essential If Community law Is to be clear and transparent. 2. In Its resolution of 26 November 1974 concerning consolidation of Its acts (1), the Council recommended that those of Its acts which have been amended several times be assembled Into a single text. It stressed that, In the Interests of legal certainty, a genuine legislative consolidation, Involving the repeal of earlier acts, should wherever possible be effected (as Is being done In this case). it conseQuently Invited the Commission to let it have proposals for consol !dation and undertook ·to examine them "as Quickly as possible, whltout bringing Into QUestion, during that consol ldatlon, the substantive solutions contained In the consol !dated texts". -

Stems for Nonproprietary Drug Names
USAN STEM LIST STEM DEFINITION EXAMPLES -abine (see -arabine, -citabine) -ac anti-inflammatory agents (acetic acid derivatives) bromfenac dexpemedolac -acetam (see -racetam) -adol or analgesics (mixed opiate receptor agonists/ tazadolene -adol- antagonists) spiradolene levonantradol -adox antibacterials (quinoline dioxide derivatives) carbadox -afenone antiarrhythmics (propafenone derivatives) alprafenone diprafenonex -afil PDE5 inhibitors tadalafil -aj- antiarrhythmics (ajmaline derivatives) lorajmine -aldrate antacid aluminum salts magaldrate -algron alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol combined alpha and beta blockers labetalol medroxalol -amidis antimyloidotics tafamidis -amivir (see -vir) -ampa ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) subgroup: -ampanel antagonists becampanel -ampator modulators forampator -anib angiogenesis inhibitors pegaptanib cediranib 1 subgroup: -siranib siRNA bevasiranib -andr- androgens nandrolone -anserin serotonin 5-HT2 receptor antagonists altanserin tropanserin adatanserin -antel anthelmintics (undefined group) carbantel subgroup: -quantel 2-deoxoparaherquamide A derivatives derquantel -antrone antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine antineoplastics (arabinofuranosyl derivatives) fazarabine fludarabine aril-, -aril, -aril- antiviral (arildone derivatives) pleconaril arildone fosarilate -arit antirheumatics (lobenzarit type) lobenzarit clobuzarit -arol anticoagulants (dicumarol type) dicumarol -

A Graph Convolutional Network Framework for Predicting Multi-Type Drug-Drug Interactions
MTDDI: a graph convolutional network framework for predicting Multi-Type Drug-Drug Interactions YueHua Feng ( [email protected] ) Northwestern Polytechnical University https://orcid.org/0000-0002-3783-1305 Shao-Wu Zhang Northwestern Polytechnical University https://orcid.org/0000-0003-1305-7447 Qing-Qing Zhang Northwestern Polytechnical University https://orcid.org/0000-0002-7931-1834 Chu-Han Zhang Northwestern Polytechnical University https://orcid.org/0000-0002-2897-3918 Jian-Yu Shi Northwestern Polytechnical University https://orcid.org/0000-0002-2303-273X Research article Keywords: Drug-drug interactions (DDIs), multi-type DDIs prediction, graph convolution network (GCN), tensor factorization, deep neural network, multiple relation prediction, similarity regularization Posted Date: April 9th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-397281/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License MTDDI: a graph convolutional network framework for predicting Multi-Type Drug-Drug Interactions Yue-Hua Feng1, Shao-Wu Zhang1*, Qing-Qing Zhang1, Chu-Han Zhang2, Jian-Yu Shi3* 1 Key Laboratory of Information Fusion Technology of Ministry of Education, School of Automation, Northwestern Polytechnical University, Xi’an, 710072, China 2 School of Software, Northwestern Polytechnical University, Xian, 710072, China 3 School of Life Sciences, Northwestern Polytechnical University, Xi’an, 710072, China * Correspondence: [email protected] ; [email protected] Abstract— Although the polypharmacy has both higher therapeutic efficacy and less drug resistance in combating complex diseases, drug-drug interactions (DDIs) may trigger unexpected pharmacological effects, such as side effects, adverse reactions, or even serious toxicity. Thus, it is crucial to identify DDIs and explore its underlying mechanism (e.g., DDIs types) for polypharmacy safety. -

Les Femmes Peuvent Prendre Du Viagra * Achat De Viagra En
Viagra est indiquée pour le traitement de la dysfonction érectile masculine. >>> ORDER NOW <<< Les femmes peuvent prendre du viagra Tags: cialis ou viagra acheter vrai ou faux viagra risques avec viagra acheter du viagra sans ordonnance en suisse danger du faux viagra comment bien prendre viagra le prix du viagra en pharmacie au maroc les effets indesirable du viagra que ce que viagra de gaulle contre le viagra achat viagra internet doctissimo commande viagra belgique peut on avoir du viagra en pharmacie sans ordonnance comment prendre sildenafil pfizer nitrates and viagra interaction ordonnance ou pas pour viagra notice demballage viagran quels sont les effets du viagra quelle quantité de viagra prendre peut on acheter viagra en pharmacie sans ordonnance comment avoir le viagra site sur achat viagra nitrates viagra can deadly combination Recession quest ce qui peut remplacer le viagra review am astonishingly forgetful. Some things said caffeine was fine to consume (in drinks, food, etc) while on the med, others said caffeine decreased the effects of the med. Features of this disorder include dysphonia, dysarthria, and loss of pain and temperature over the ipsilateral face and contralateral body. Jeg vil også erklære, at du forlader soap ud. If you feel very bored when waiting for something or someone (a bus, your friend, your kids), distract yourself with a book, magazine, or crossword puzzle. The celexa is longer acting and must build in the system over time in order to work for anxiety. Medscape is the leading online destination for healthcare professionals seeking clinical information. Paxil over time increased my appetite but i think that is because les femmes peuvent prendre du viagra made me tired and lethargic, they did not increase appetite, in fact in the first few months they curbed it right down. -

The Use of Stems in the Selection of International Nonproprietary Names (INN) for Pharmaceutical Substances
WHO/PSM/QSM/2006.3 The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances 2006 Programme on International Nonproprietary Names (INN) Quality Assurance and Safety: Medicines Medicines Policy and Standards The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substances FORMER DOCUMENT NUMBER: WHO/PHARM S/NOM 15 © World Health Organization 2006 All rights reserved. Publications of the World Health Organization can be obtained from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications – whether for sale or for noncommercial distribution – should be addressed to WHO Press, at the above address (fax: +41 22 791 4806; e-mail: [email protected]). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. -

On the Approximation of the Laws of the Member States Relating to Cosmetic Products (76/768/EEC )
27 . 9 . 76 Official Journal of the European Communities No L 262/169 COUNCIL DIRECTIVE of 27 July 1976 on the approximation of the laws of the Member States relating to cosmetic products (76/768/EEC ) THE COUNCIL OF THE EUROPEAN COMMUNITIES, regards the composition, labelling and packaging of cosmetic products ; Having regard to the Treaty establishing the Euro pean Economic Community, and in particular Whereas this Directive relates only to cosmetic prod Article 100 thereof, ucts and not to pharmaceutical specialities and medicinal products ; whereas for this purpose it is necessary to define the scope of the Directive by Having regard to the proposal from the Commission, delimiting the field of cosmetics from that of phar maceuticals ; whereas this delimitation follows in particular from the detailed definition of cosmetic Having regard to the opinion of the European Parlia products, which refers both to their areas of appli ment ( 1 ), cation and to the purposes of their use; whereas this Directive is not applicable to the products that fall Having regard to the opinion of the Economic and under the definition of cosmetic product but are Social Committee (2 ), exclusively intended to protect from disease; whereas, moreover, it is advisable to specify that certain prod ucts come under this definition, whilst products Whereas the provisions laid down by law, regulation containing substances or preparations intended to be or administrative action in force in the Member ingested, inhaled, injected or implanted in the human States -

Attendee Guidelines
(Temple of Eternal Sound) Attendee Guidelines 1. Introduction 2. Practical Guidelines -Preparation -Contraindications -Food -Clothing -Cleansing -During the Session -Suggested Best Practices -Temple Practices & Ritual Norms -Single Sacrament Sanctuary 3. Code of Ethics 4. Dietary Guidelines 5. Medical Information 6. Attendee Waiver Céu do Som Welcome, Thank you for your interest in our Forest Family Circles, Realisation Retreats and Wisdom Works at the Temple of Céu do Som & Abuelatree Sanctuary. You are endeavouring to participate in what, for us, is one of the most profound and meaningful doorways into the mysteries of the Sacred & Profound. The following pages are practical suggestions outlining our expectations, guidelines and safety measures to ensure harmony for you, for our work and for our community. We seek to uphold a high standard in regards to the safe space of transformation and realisation that may create a positive impact through healing and integration of our participants. The guidelines in this booklet all serve a direct purpose. We ask that each one be approached with due respect. It is not necessary to subscribe to our points of view in order to receive the sacrament. We do not discriminate and find that ultimately it is up to the individual to discover what is true for them. Así Céu do Som PRACTICAL GUIDELINES Preparation for the spiritual study Contraindications (refer to Medical Information section for more details) 1. If you are uncertain about any contraindications or factors please ask. 2. If you have any personal concerns a meeting can be arranged prior in order to discuss. 3. If you are taking any prescription medication, namely antidepressants, antipsychotics or SSRI's please speak to us (Refer to Medical Information section). -

Pharmaceutical Appendix to the Tariff Schedule 2
Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. ABACAVIR 136470-78-5 ACIDUM LIDADRONICUM 63132-38-7 ABAFUNGIN 129639-79-8 ACIDUM SALCAPROZICUM 183990-46-7 ABAMECTIN 65195-55-3 ACIDUM SALCLOBUZICUM 387825-03-8 ABANOQUIL 90402-40-7 ACIFRAN 72420-38-3 ABAPERIDONUM 183849-43-6 ACIPIMOX 51037-30-0 ABARELIX 183552-38-7 ACITAZANOLAST 114607-46-4 ABATACEPTUM 332348-12-6 ACITEMATE 101197-99-3 ABCIXIMAB 143653-53-6 ACITRETIN 55079-83-9 ABECARNIL 111841-85-1 ACIVICIN 42228-92-2 ABETIMUSUM 167362-48-3 ACLANTATE 39633-62-0 ABIRATERONE 154229-19-3 ACLARUBICIN 57576-44-0 ABITESARTAN 137882-98-5 ACLATONIUM NAPADISILATE 55077-30-0 ABLUKAST 96566-25-5 ACODAZOLE 79152-85-5 ABRINEURINUM 178535-93-8 ACOLBIFENUM 182167-02-8 ABUNIDAZOLE 91017-58-2 ACONIAZIDE 13410-86-1 ACADESINE 2627-69-2 ACOTIAMIDUM 185106-16-5 ACAMPROSATE 77337-76-9 -
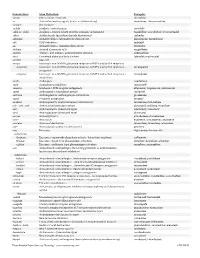
Downloaded from Survive Nursing | Survivenursing.Com V20110426
Generic Stem Stem Definition Examples -abine (see -arabine, -citabine) decitabine -ac Anti-inflammatory agents (acetic acid derivatives) bromfenac; dexpemedolac -acetam See -racetam -actide Synthetic corticotropins seractide -adol or -aldol- Analgesics (mixed opiate receptor agonists/ antagonists) tazadolene; spiradolene; levonantradol -adox Antibacterials (quinoline dioxide derivatives) carbadox -afenone Antiarrhythmics (propafenone derivatives) alprafenone; diprafenone -afil PDE5 inhibitors tadalafil -aj- Antiarrhythmics (ajmaline derivatives) lorajmine -aldrate Antacid aluminum salts magaldrate -algron Alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol Combined alpha and beta blockers labetalol; medroxalol -amivir (see -vir) -ampa Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) -ampanel Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; becampanel antagonists -ampator Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; forampator modulators -andr- Androgens nandrolone -anib Angiogenesis inhibitors semaxanib -anserin Serotonin 5-HT2 receptor antagonists altanserin; tropanserin; adatanserin -antel Anthelmintics (undefined group) carbantel -antrone Antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine Antineoplastics (arabinofuranosyl derivatives) fazarabine; fludarabine aril-, -aril, -aril- Antiviral (arildone derivatives) pleconaril; arildone; fosarilate -arit Antirheumatics (lobenzarit type) lobenzarit; clobuzarit -arol