3.3 Convex Polyhedron
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hardware Support for Non-Photorealistic Rendering
Hardware Support for Non-photorealistic Rendering Ramesh Raskar MERL, Mitsubishi Electric Research Labs (ii) Abstract (i) Special features such as ridges, valleys and silhouettes, of a polygonal scene are usually displayed by explicitly identifying and then rendering ‘edges’ for the corresponding geometry. The candidate edges are identified using the connectivity information, which requires preprocessing of the data. We present a non- obvious but surprisingly simple to implement technique to render such features without connectivity information or preprocessing. (iii) (iv) At the hardware level, based only on the vertices of a given flat polygon, we introduce new polygons, with appropriate color, shape and orientation, so that they eventually appear as special features. 1 INTRODUCTION Figure 1: (i) Silhouettes, (ii) ridges, (iii) valleys and (iv) their combination for a polygonal 3D model rendered by processing Sharp features convey a great deal of information with very few one polygon at a time. strokes. Technical illustrations, engineering CAD diagrams as well as non-photo-realistic rendering techniques exploit these features to enhance the appearance of the underlying graphics usually not supported by rendering APIs or hardware. The models. The most commonly used features are silhouettes, creases rendering hardware is typically more suited to working on a small and intersections. For polygonal meshes, the silhouette edges amount of data at a time. For example, most pipelines accept just consists of visible segments of all edges that connect back-facing a sequence of triangles (or triangle soups) and all the information polygons to front-facing polygons. A crease edge is a ridge if the necessary for rendering is contained in the individual triangles. -

Arc Length. Surface Area
Calculus 2 Lia Vas Arc Length. Surface Area. Arc Length. Suppose that y = f(x) is a continuous function with a continuous derivative on [a; b]: The arc length L of f(x) for a ≤ x ≤ b can be obtained by integrating the length element ds from a to b: The length element ds on a sufficiently small interval can be approximated by the hypotenuse of a triangle with sides dx and dy: p Thus ds2 = dx2 + dy2 ) ds = dx2 + dy2 and so s s Z b Z b q Z b dy2 Z b dy2 L = ds = dx2 + dy2 = (1 + )dx2 = (1 + )dx: a a a dx2 a dx2 2 dy2 dy 0 2 Note that dx2 = dx = (y ) : So the formula for the arc length becomes Z b q L = 1 + (y0)2 dx: a Area of a surface of revolution. Suppose that y = f(x) is a continuous function with a continuous derivative on [a; b]: To compute the surface area Sx of the surface obtained by rotating f(x) about x-axis on [a; b]; we can integrate the surface area element dS which can be approximated as the product of the circumference 2πy of the circle with radius y and the height that is given by q the arc length element ds: Since ds is 1 + (y0)2dx; the formula that computes the surface area is Z b q 0 2 Sx = 2πy 1 + (y ) dx: a If y = f(x) is rotated about y-axis on [a; b]; then dS is the product of the circumference 2πx of the circle with radius x and the height that is given by the arc length element ds: Thus, the formula that computes the surface area is Z b q 0 2 Sy = 2πx 1 + (y ) dx: a Practice Problems. -

Cones, Pyramids and Spheres
The Improving Mathematics Education in Schools (TIMES) Project MEASUREMENT AND GEOMETRY Module 12 CONES, PYRAMIDS AND SPHERES A guide for teachers - Years 9–10 June 2011 YEARS 910 Cones, Pyramids and Spheres (Measurement and Geometry : Module 12) For teachers of Primary and Secondary Mathematics 510 Cover design, Layout design and Typesetting by Claire Ho The Improving Mathematics Education in Schools (TIMES) Project 2009‑2011 was funded by the Australian Government Department of Education, Employment and Workplace Relations. The views expressed here are those of the author and do not necessarily represent the views of the Australian Government Department of Education, Employment and Workplace Relations. © The University of Melbourne on behalf of the International Centre of Excellence for Education in Mathematics (ICE‑EM), the education division of the Australian Mathematical Sciences Institute (AMSI), 2010 (except where otherwise indicated). This work is licensed under the Creative Commons Attribution‑NonCommercial‑NoDerivs 3.0 Unported License. 2011. http://creativecommons.org/licenses/by‑nc‑nd/3.0/ The Improving Mathematics Education in Schools (TIMES) Project MEASUREMENT AND GEOMETRY Module 12 CONES, PYRAMIDS AND SPHERES A guide for teachers - Years 9–10 June 2011 Peter Brown Michael Evans David Hunt Janine McIntosh Bill Pender Jacqui Ramagge YEARS 910 {4} A guide for teachers CONES, PYRAMIDS AND SPHERES ASSUMED KNOWLEDGE • Familiarity with calculating the areas of the standard plane figures including circles. • Familiarity with calculating the volume of a prism and a cylinder. • Familiarity with calculating the surface area of a prism. • Facility with visualizing and sketching simple three‑dimensional shapes. • Facility with using Pythagoras’ theorem. • Facility with rounding numbers to a given number of decimal places or significant figures. -

Lateral and Surface Area of Right Prisms 1 Jorge Is Trying to Wrap a Present That Is in a Box Shaped As a Right Prism
CHAPTER 11 You will need Lateral and Surface • a ruler A • a calculator Area of Right Prisms c GOAL Calculate lateral area and surface area of right prisms. Learn about the Math A prism is a polyhedron (solid whose faces are polygons) whose bases are congruent and parallel. When trying to identify a right prism, ask yourself if this solid could have right prism been created by placing many congruent sheets of paper on prism that has bases aligned one above the top of each other. If so, this is a right prism. Some examples other and has lateral of right prisms are shown below. faces that are rectangles Triangular prism Rectangular prism The surface area of a right prism can be calculated using the following formula: SA 5 2B 1 hP, where B is the area of the base, h is the height of the prism, and P is the perimeter of the base. The lateral area of a figure is the area of the non-base faces lateral area only. When a prism has its bases facing up and down, the area of the non-base faces of a figure lateral area is the area of the vertical faces. (For a rectangular prism, any pair of opposite faces can be bases.) The lateral area of a right prism can be calculated by multiplying the perimeter of the base by the height of the prism. This is summarized by the formula: LA 5 hP. Copyright © 2009 by Nelson Education Ltd. Reproduction permitted for classrooms 11A Lateral and Surface Area of Right Prisms 1 Jorge is trying to wrap a present that is in a box shaped as a right prism. -

Regular Polyhedra of Index 2
REGULAR POLYHEDRA OF INDEX 2 A dissertation presented by Anthony Cutler to The Department of Mathematics In partial fulfillment of the requirements for the degree of Doctor of Philosophy in the field of Mathematics Northeastern University Boston, Massachusetts April, 2009 1 REGULAR POLYHEDRA OF INDEX 2 by Anthony Cutler ABSTRACT OF DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Mathematics in the Graduate School of Arts and Sciences of Northeastern University, April 2009 2 We classify all finite regular polyhedra of index 2, as defined in Section 2 herein. The definition requires the polyhedra to be combinatorially flag transitive, but does not require them to have planar or convex faces or vertex-figures, and neither does it require the polyhedra to be orientable. We find there are 10 combinatorially regular polyhedra of index 2 with vertices on one orbit, and 22 infinite families of combinatorially regular polyhedra of index 2 with vertices on two orbits, where polyhedra in the same family differ only in the relative diameters of their vertex orbits. For each such polyhedron, or family of polyhedra, we provide the underlying map, as well as a geometric diagram showing a representative face for each face orbit, and a verification of the polyhedron’s combinatorial regularity. A self-contained completeness proof is given. Exactly five of the polyhedra have planar faces, which is consistent with a previously known result. We conclude by describing a non-Petrie duality relation among regular polyhedra of index 2, and suggest how it can be extended to other combinatorially regular polyhedra. -

Systematics of Atomic Orbital Hybridization of Coordination Polyhedra: Role of F Orbitals
molecules Article Systematics of Atomic Orbital Hybridization of Coordination Polyhedra: Role of f Orbitals R. Bruce King Department of Chemistry, University of Georgia, Athens, GA 30602, USA; [email protected] Academic Editor: Vito Lippolis Received: 4 June 2020; Accepted: 29 June 2020; Published: 8 July 2020 Abstract: The combination of atomic orbitals to form hybrid orbitals of special symmetries can be related to the individual orbital polynomials. Using this approach, 8-orbital cubic hybridization can be shown to be sp3d3f requiring an f orbital, and 12-orbital hexagonal prismatic hybridization can be shown to be sp3d5f2g requiring a g orbital. The twists to convert a cube to a square antiprism and a hexagonal prism to a hexagonal antiprism eliminate the need for the highest nodality orbitals in the resulting hybrids. A trigonal twist of an Oh octahedron into a D3h trigonal prism can involve a gradual change of the pair of d orbitals in the corresponding sp3d2 hybrids. A similar trigonal twist of an Oh cuboctahedron into a D3h anticuboctahedron can likewise involve a gradual change in the three f orbitals in the corresponding sp3d5f3 hybrids. Keywords: coordination polyhedra; hybridization; atomic orbitals; f-block elements 1. Introduction In a series of papers in the 1990s, the author focused on the most favorable coordination polyhedra for sp3dn hybrids, such as those found in transition metal complexes. Such studies included an investigation of distortions from ideal symmetries in relatively symmetrical systems with molecular orbital degeneracies [1] In the ensuing quarter century, interest in actinide chemistry has generated an increasing interest in the involvement of f orbitals in coordination chemistry [2–7]. -

Binary Icosahedral Group and 600-Cell
Article Binary Icosahedral Group and 600-Cell Jihyun Choi and Jae-Hyouk Lee * Department of Mathematics, Ewha Womans University 52, Ewhayeodae-gil, Seodaemun-gu, Seoul 03760, Korea; [email protected] * Correspondence: [email protected]; Tel.: +82-2-3277-3346 Received: 10 July 2018; Accepted: 26 July 2018; Published: 7 August 2018 Abstract: In this article, we have an explicit description of the binary isosahedral group as a 600-cell. We introduce a method to construct binary polyhedral groups as a subset of quaternions H via spin map of SO(3). In addition, we show that the binary icosahedral group in H is the set of vertices of a 600-cell by applying the Coxeter–Dynkin diagram of H4. Keywords: binary polyhedral group; icosahedron; dodecahedron; 600-cell MSC: 52B10, 52B11, 52B15 1. Introduction The classification of finite subgroups in SLn(C) derives attention from various research areas in mathematics. Especially when n = 2, it is related to McKay correspondence and ADE singularity theory [1]. The list of finite subgroups of SL2(C) consists of cyclic groups (Zn), binary dihedral groups corresponded to the symmetry group of regular 2n-gons, and binary polyhedral groups related to regular polyhedra. These are related to the classification of regular polyhedrons known as Platonic solids. There are five platonic solids (tetrahedron, cubic, octahedron, dodecahedron, icosahedron), but, as a regular polyhedron and its dual polyhedron are associated with the same symmetry groups, there are only three binary polyhedral groups(binary tetrahedral group 2T, binary octahedral group 2O, binary icosahedral group 2I) related to regular polyhedrons. -
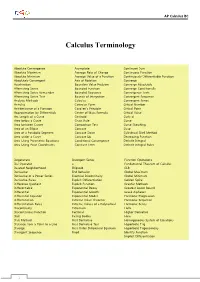
Calculus Terminology
AP Calculus BC Calculus Terminology Absolute Convergence Asymptote Continued Sum Absolute Maximum Average Rate of Change Continuous Function Absolute Minimum Average Value of a Function Continuously Differentiable Function Absolutely Convergent Axis of Rotation Converge Acceleration Boundary Value Problem Converge Absolutely Alternating Series Bounded Function Converge Conditionally Alternating Series Remainder Bounded Sequence Convergence Tests Alternating Series Test Bounds of Integration Convergent Sequence Analytic Methods Calculus Convergent Series Annulus Cartesian Form Critical Number Antiderivative of a Function Cavalieri’s Principle Critical Point Approximation by Differentials Center of Mass Formula Critical Value Arc Length of a Curve Centroid Curly d Area below a Curve Chain Rule Curve Area between Curves Comparison Test Curve Sketching Area of an Ellipse Concave Cusp Area of a Parabolic Segment Concave Down Cylindrical Shell Method Area under a Curve Concave Up Decreasing Function Area Using Parametric Equations Conditional Convergence Definite Integral Area Using Polar Coordinates Constant Term Definite Integral Rules Degenerate Divergent Series Function Operations Del Operator e Fundamental Theorem of Calculus Deleted Neighborhood Ellipsoid GLB Derivative End Behavior Global Maximum Derivative of a Power Series Essential Discontinuity Global Minimum Derivative Rules Explicit Differentiation Golden Spiral Difference Quotient Explicit Function Graphic Methods Differentiable Exponential Decay Greatest Lower Bound Differential -

Enhancing Strategic Discourse Systematically Using Climate Metaphors Widespread Comprehension of System Dynamics in Weather Patterns As a Resource -- /
Alternative view of segmented documents via Kairos 24 August 2015 | Draft Enhancing Strategic Discourse Systematically using Climate Metaphors Widespread comprehension of system dynamics in weather patterns as a resource -- / -- Introduction Systematic global insight of memorable quality? Towards memorable framing of global climate of governance processes? Visual representations of globality of requisite variety for global governance Four-dimensional requisite for a time-bound global civilization? Comprehending the shapes of time through four-dimensional uniform polychora Five-fold ordering of strategic engagement with time Interplay of cognitive patterns in discourse on systemic change Five-fold cognitive dynamics of relevance to governance? Decision-making capacity versus Distinction-making capacity: embodying whether as weather From star-dom to whizdom to isdom? From space-ship design to time-ship embodiment as a requisite metaphor of governance References Prepared in anticipation of the United Nations Climate Change Conference (Paris, 2015) Introduction There is considerable familiarity with the dynamics of climate and weather through the seasons and in different locations. These provide a rich source of metaphor, widely shared, and frequently exploited as a means of communicating subtle insights into social phenomena, strategic options and any resistance to them. It is however now difficult to claim any coherence to the strategic discourse on which humanity and global governance is held to be dependent. This is evident in the deprecation of one political faction by another, the exacerbation of conflictual perceptions by the media, the daily emergence of intractable crises, and the contradictory assertions of those claiming expertise in one arena or another. The dynamics have been caricatured as blame-gaming, as separately discussed (Blame game? It's them not us ! 2015). -

A Tourist Guide to the RCSR
A tourist guide to the RCSR Some of the sights, curiosities, and little-visited by-ways Michael O'Keeffe, Arizona State University RCSR is a Reticular Chemistry Structure Resource available at http://rcsr.net. It is open every day of the year, 24 hours a day, and admission is free. It consists of data for polyhedra and 2-periodic and 3-periodic structures (nets). Visitors unfamiliar with the resource are urged to read the "about" link first. This guide assumes you have. The guide is designed to draw attention to some of the attractions therein. If they sound particularly attractive please visit them. It can be a nice way to spend a rainy Sunday afternoon. OKH refers to M. O'Keeffe & B. G. Hyde. Crystal Structures I: Patterns and Symmetry. Mineral. Soc. Am. 1966. This is out of print but due as a Dover reprint 2019. POLYHEDRA Read the "about" for hints on how to use the polyhedron data to make accurate drawings of polyhedra using crystal drawing programs such as CrystalMaker (see "links" for that program). Note that they are Cartesian coordinates for (roughly) equal edge. To make the drawing with unit edge set the unit cell edges to all 10 and divide the coordinates given by 10. There seems to be no generally-agreed best embedding for complex polyhedra. It is generally not possible to have equal edge, vertices on a sphere and planar faces. Keywords used in the search include: Simple. Each vertex is trivalent (three edges meet at each vertex) Simplicial. Each face is a triangle. -

Four-Dimensional Regular Polytopes
faculty of science mathematics and applied and engineering mathematics Four-dimensional regular polytopes Bachelor’s Project Mathematics November 2020 Student: S.H.E. Kamps First supervisor: Prof.dr. J. Top Second assessor: P. Kiliçer, PhD Abstract Since Ancient times, Mathematicians have been interested in the study of convex, regular poly- hedra and their beautiful symmetries. These five polyhedra are also known as the Platonic Solids. In the 19th century, the four-dimensional analogues of the Platonic solids were described mathe- matically, adding one regular polytope to the collection with no analogue regular polyhedron. This thesis describes the six convex, regular polytopes in four-dimensional Euclidean space. The focus lies on deriving information about their cells, faces, edges and vertices. Besides that, the symmetry groups of the polytopes are touched upon. To better understand the notions of regularity and sym- metry in four dimensions, our journey begins in three-dimensional space. In this light, the thesis also works out the details of a proof of prof. dr. J. Top, showing there exist exactly five convex, regular polyhedra in three-dimensional space. Keywords: Regular convex 4-polytopes, Platonic solids, symmetry groups Acknowledgements I would like to thank prof. dr. J. Top for supervising this thesis online and adapting to the cir- cumstances of Covid-19. I also want to thank him for his patience, and all his useful comments in and outside my LATEX-file. Also many thanks to my second supervisor, dr. P. Kılıçer. Furthermore, I would like to thank Jeanne for all her hospitality and kindness to welcome me in her home during the process of writing this thesis. -

Regular Polyhedra Through Time
Fields Institute I. Hubard Polytopes, Maps and their Symmetries September 2011 Regular polyhedra through time The greeks were the first to study the symmetries of polyhedra. Euclid, in his Elements showed that there are only five regular solids (that can be seen in Figure 1). In this context, a polyhe- dron is regular if all its polygons are regular and equal, and you can find the same number of them at each vertex. Figure 1: Platonic Solids. It is until 1619 that Kepler finds other two regular polyhedra: the great dodecahedron and the great icosahedron (on Figure 2. To do so, he allows \false" vertices and intersection of the (convex) faces of the polyhedra at points that are not vertices of the polyhedron, just as the I. Hubard Polytopes, Maps and their Symmetries Page 1 Figure 2: Kepler polyhedra. 1619. pentagram allows intersection of edges at points that are not vertices of the polygon. In this way, the vertex-figure of these two polyhedra are pentagrams (see Figure 3). Figure 3: A regular convex pentagon and a pentagram, also regular! In 1809 Poinsot re-discover Kepler's polyhedra, and discovers its duals: the small stellated dodecahedron and the great stellated dodecahedron (that are shown in Figure 4). The faces of such duals are pentagrams, and are organized on a \convex" way around each vertex. Figure 4: The other two Kepler-Poinsot polyhedra. 1809. A couple of years later Cauchy showed that these are the only four regular \star" polyhedra. We note that the convex hull of the great dodecahedron, great icosahedron and small stellated dodecahedron is the icosahedron, while the convex hull of the great stellated dodecahedron is the dodecahedron.