The Identification of Novel Regions for Reproduction Trait in Landrace and Large White Pigs Using a Single Step Genome-Wide Association Study
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Oxidative Stress-Induced Chromosome Breaks Within
Tan et al. Human Genomics (2018) 12:29 https://doi.org/10.1186/s40246-018-0160-8 PRIMARY RESEARCH Open Access Oxidative stress-induced chromosome breaks within the ABL gene: a model for chromosome rearrangement in nasopharyngeal carcinoma Sang-Nee Tan1, Sai-Peng Sim1* and Alan Soo-Beng Khoo2 Abstract Background: The mechanism underlying chromosome rearrangement in nasopharyngeal carcinoma (NPC) remains elusive. It is known that most of the aetiological factors of NPC trigger oxidative stress. Oxidative stress is a potent apoptotic inducer. During apoptosis, chromatin cleavage and DNA fragmentation occur. However, cells may undergo DNA repair and survive apoptosis. Non-homologous end joining (NHEJ) pathway has been known as the primary DNA repair system in human cells. The NHEJ process may repair DNA ends without any homology, although region of microhomology (a few nucleotides) is usually utilised by this DNA repair system. Cells that evade apoptosis via erroneous DNA repair may carry chromosomal aberration. Apoptotic nuclease was found to be associated with nuclear matrix during apoptosis. Matrix association region/scaffold attachment region (MAR/SAR) is the binding site of the chromosomal DNA loop structure to the nuclear matrix. When apoptotic nuclease is associated with nuclear matrix during apoptosis, it potentially cleaves at MAR/SAR. Cells that survive apoptosis via compromised DNA repair may carry chromosome rearrangement contributing to NPC tumourigenesis. The Abelson murine leukaemia (ABL) gene at 9q34 was targeted in this study as 9q34 is a common region of loss in NPC. This study aimed to identify the chromosome breakages and/or rearrangements in the ABL gene in cells undergoing oxidative stress-induced apoptosis. -

Ageing-Associated Changes in DNA Methylation in X and Y Chromosomes
Kananen and Marttila Epigenetics & Chromatin (2021) 14:33 Epigenetics & Chromatin https://doi.org/10.1186/s13072-021-00407-6 RESEARCH Open Access Ageing-associated changes in DNA methylation in X and Y chromosomes Laura Kananen1,2,3,4* and Saara Marttila4,5* Abstract Background: Ageing displays clear sexual dimorphism, evident in both morbidity and mortality. Ageing is also asso- ciated with changes in DNA methylation, but very little focus has been on the sex chromosomes, potential biological contributors to the observed sexual dimorphism. Here, we sought to identify DNA methylation changes associated with ageing in the Y and X chromosomes, by utilizing datasets available in data repositories, comprising in total of 1240 males and 1191 females, aged 14–92 years. Results: In total, we identifed 46 age-associated CpG sites in the male Y, 1327 age-associated CpG sites in the male X, and 325 age-associated CpG sites in the female X. The X chromosomal age-associated CpGs showed signifcant overlap between females and males, with 122 CpGs identifed as age-associated in both sexes. Age-associated X chro- mosomal CpGs in both sexes were enriched in CpG islands and depleted from gene bodies and showed no strong trend towards hypermethylation nor hypomethylation. In contrast, the Y chromosomal age-associated CpGs were enriched in gene bodies, and showed a clear trend towards hypermethylation with age. Conclusions: Signifcant overlap in X chromosomal age-associated CpGs identifed in males and females and their shared features suggest that despite the uneven chromosomal dosage, diferences in ageing-associated DNA methylation changes in the X chromosome are unlikely to be a major contributor of sex dimorphism in ageing. -
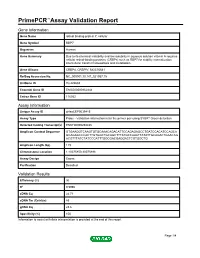
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name retinol binding protein 7, cellular Gene Symbol RBP7 Organism Human Gene Summary Due to its chemical instability and low solubility in aqueous solution vitamin A requires cellular retinol-binding proteins (CRBPs) such as RBP7 for stability internalization intercellular transfer homeostasis and metabolism. Gene Aliases CRBP4, CRBPIV, MGC70641 RefSeq Accession No. NC_000001.10, NT_021937.19 UniGene ID Hs.422688 Ensembl Gene ID ENSG00000162444 Entrez Gene ID 116362 Assay Information Unique Assay ID qHsaCEP0039415 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000294435 Amplicon Context Sequence GTGAAGGTCAAGTGTGCAAACAGACATTCCAGAGAGCCTGATCCACATCCAGCA GCAGAGCCCACTTGTGGCTGCAGCTTTATGCCAAATTATATTGCAGACTGAACAG ACGTTTATCTATCCCATTTGGCGACGAGGACTCGTGGCTG Amplicon Length (bp) 119 Chromosome Location 1:10075850-10075998 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 96 R2 0.9996 cDNA Cq 24.71 cDNA Tm (Celsius) 83 gDNA Cq 23.6 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report RBP7, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -

Causal Varian Discovery in Familial Congenital Heart Disease - an Integrative -Omic Approach Wendy Demos Marquette University
Marquette University e-Publications@Marquette Master's Theses (2009 -) Dissertations, Theses, and Professional Projects Causal Varian discovery in Familial Congenital Heart Disease - An Integrative -Omic Approach Wendy Demos Marquette University Recommended Citation Demos, Wendy, "Causal Varian discovery in Familial Congenital Heart Disease - An Integrative -Omic Approach" (2012). Master's Theses (2009 -). 140. https://epublications.marquette.edu/theses_open/140 CAUSAL VARIANT DISCOVERY IN FAMILIAL CONGENITAL HEART DISEASE – AN INTEGRATIVE –OMIC APPROACH by Wendy M. Demos A Thesis submitted to the Faculty of the Graduate School, Marquette University, in Partial Fulfillment of the Requirements for the Degree of Master of Science Milwaukee, Wisconsin May 2012 ABSTRACT CAUSAL VARIANT DISCOVERY IN FAMILIAL CONGENITAL HEART DISEASE – AN INTEGRATIVE –OMIC APPROACH Wendy M. Demos Marquette University, 2012 Background : Hypoplastic left heart syndrome (HLHS) is a congenital heart defect that leads to neonatal death or compromised quality of life for those affected and their families. This syndrome requires extensive medical intervention for the affected to survive. It is characterized by significant underdevelopment or non-existence of the components of the left heart and the aorta, including the left ventricular cavity and mass. There are many factors ranging from genetics to environmental relationships hypothesized to lead to the development of the syndrome, including recent studies suggesting a link between hearing impairment and congenital heart defects (CHD). Although broadly characterized those factors remain poorly understood. The goal of this project is to systematically utilize bioinformatics tools to determine the relationships of novel mutations found in exome sequencing to a familial congenital heart defect. Methods A systematic genomic and proteomic approach involving exome sequencing, pathway analysis, and protein modeling was implemented to examine exome sequencing data of a patient with HLHS. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

Slitrks Control Excitatory and Inhibitory Synapse Formation with LAR
Slitrks control excitatory and inhibitory synapse SEE COMMENTARY formation with LAR receptor protein tyrosine phosphatases Yeong Shin Yima,1, Younghee Kwonb,1, Jungyong Namc, Hong In Yoona, Kangduk Leeb, Dong Goo Kima, Eunjoon Kimc, Chul Hoon Kima,2, and Jaewon Kob,2 aDepartment of Pharmacology, Brain Research Institute, Brain Korea 21 Project for Medical Science, Severance Biomedical Science Institute, Yonsei University College of Medicine, Seoul 120-752, Korea; bDepartment of Biochemistry, College of Life Science and Biotechnology, Yonsei University, Seoul 120-749, Korea; and cCenter for Synaptic Brain Dysfunctions, Institute for Basic Science, Department of Biological Sciences, Korea Advanced Institute of Science and Technology, Daejeon 305-701, Korea Edited by Thomas C. Südhof, Stanford University School of Medicine, Stanford, CA, and approved December 26, 2012 (received for review June 11, 2012) The balance between excitatory and inhibitory synaptic inputs, share a similar domain organization comprising three Ig domains which is governed by multiple synapse organizers, controls neural and four to eight fibronectin type III repeats. LAR-RPTP family circuit functions and behaviors. Slit- and Trk-like proteins (Slitrks) are members are evolutionarily conserved and are functionally required a family of synapse organizers, whose emerging synaptic roles are for axon guidance and synapse formation (15). Recent studies have incompletely understood. Here, we report that Slitrks are enriched shown that netrin-G ligand-3 (NGL-3), neurotrophin receptor ty- in postsynaptic densities in rat brains. Overexpression of Slitrks rosine kinase C (TrkC), and IL-1 receptor accessory protein-like 1 promoted synapse formation, whereas RNAi-mediated knock- (IL1RAPL1) bind to all three LAR-RPTP family members or dis- down of Slitrks decreased synapse density. -

Role of Ube4b in the Ubiquitination of the Htlv-1 Tax Oncoprotein and Nf-B Activation
ROLE OF UBE4B IN THE UBIQUITINATION OF THE HTLV-1 TAX ONCOPROTEIN AND NF-B ACTIVATION by Teng Han A thesis submitted to Johns Hopkins University in conformity with the requirements for the degree of Master of Science Baltimore, Maryland April, 2014 © 2014 Teng Han All Rights Reserved i ABSTRACT Human T-cell leukemia virus type 1 (HTLV-1) is the etiological agent of adult T-cell leukemia and lymphoma (ATLL), an aggressive CD4+CD25+ malignancy. The HTLV-1 genome encodes the Tax protein that plays essential regulatory roles in oncogenic transformation of T lymphocytes by deregulating different cellular pathways, most notably NF-κB. Lysine 63 (K63)-linked polyubiquitination of Tax provides an important regulatory mechanism that promotes Tax-mediated interaction with the IKK complex and activation of NF-κB. However, the E3 ligase(s) and other host proteins regulating Tax ubiquitination are currently unknown. To identify novel Tax interacting proteins that may regulate its ubiquitination we conducted a yeast two-hybrid screen using Tax as bait. This screen yielded the E3/E4 ligase ubiquitin conjugation E4 B (UBE4B) as a novel binding partner for Tax. Here, we confirmed the interaction between Tax and UBE4B in mammalian cells by co-immunoprecipitation assays and demonstrated that they co- localized in the cytoplasm by confocal microscopy. Overexpression of UBE4B specifically enhanced Tax-induced NF-κB activation, whereas knockdown of UBE4B impaired Tax-induced NF-κB activation and induction of NF-B target genes in Jurkat T cells and ATL cell lines. Although the UBE4B promoter contains putative NF-κB binding sites, its expression was not upregulated by Tax. -

X-Linked Microtubule-Associated Protein, Mid1, Regulates Axon Development
X-linked microtubule-associated protein, Mid1, regulates axon development Tingjia Lua,b,1, Renchao Chena,b,1,2, Timothy C. Coxc,d, Randal X. Moldriche, Nyoman Kurniawanf, Guohe Tana, Jo K. Perryg, Alan Ashworthg, Perry F. Bartlette,LiXua, Jing Zhanga, Bin Lua, Mingyue Wua,b, Qi Shena, Yuanyuan Liua,b, Linda J. Richardse,h, and Zhiqi Xionga,2 aInstitute of Neuroscience and State Key Laboratory of Neuroscience, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200031, China; bUniversity of Chinese Academy of Sciences, Shanghai 200031, China; cDepartment of Pediatrics, University of Washington, Seattle, WA 98105; dDepartment of Anatomy and Developmental Biology, Monash University, Clayton, Victoria 3800, Australia; eQueensland Brain Institute, fCentre for Advanced Imaging, and hSchool of Biomedical Sciences, University of Queensland, Brisbane, QLD 4072, Australia; and gBreakthrough Breast Cancer Research Centre, Institute of Cancer Research, London SW7 3RP, United Kingdom Edited by Yuh Nung Jan, Howard Hughes Medical Institute, University of California, San Francisco, CA, and approved October 8, 2013 (received for review March 25, 2013) Opitz syndrome (OS) is a genetic neurological disorder. The gene axonal growth and branch formation whereas down-regulation of responsible for the X-linked form of OS, Midline-1 (MID1), encodes Mid1 in the developing cortex accelerated callosal axon growth an E3 ubiquitin ligase that regulates the degradation of the cata- and altered the projection pattern of callosal axons. In addition, lytic subunit of protein phosphatase 2A (PP2Ac). However, how a similar defect of axon development was observed in Mid1 Mid1 functions during neural development is largely unknown. knockout (KO) mice. -

Slitrk1 Is Localized to Excitatory Synapses and Promotes Their Development Received: 30 July 2015 François Beaubien1,2,*, Reesha Raja1,2,*, Timothy E
www.nature.com/scientificreports OPEN Slitrk1 is localized to excitatory synapses and promotes their development Received: 30 July 2015 François Beaubien1,2,*, Reesha Raja1,2,*, Timothy E. Kennedy1,3, Alyson E. Fournier1,3 & Accepted: 09 May 2016 Jean-François Cloutier1,3 Published: 07 June 2016 Following the migration of the axonal growth cone to its target area, the initial axo-dendritic contact needs to be transformed into a functional synapse. This multi-step process relies on overlapping but distinct combinations of molecules that confer synaptic identity. Slitrk molecules are transmembrane proteins that are highly expressed in the central nervous system. We found that two members of the Slitrk family, Slitrk1 and Slitrk2, can regulate synapse formation between hippocampal neurons. Slitrk1 is enriched in postsynaptic fractions and is localized to excitatory synapses. Overexpression of Slitrk1 and Slitrk2 in hippocampal neurons increased the number of synaptic contacts on these neurons. Furthermore, decreased expression of Slitrk1 in hippocampal neurons led to a reduction in the number of excitatory, but not inhibitory, synapses formed in hippocampal neuron cultures. In addition, we demonstrate that different leucine rich repeat domains of the extracellular region of Slitrk1 are necessary to mediate interactions with Slitrk binding partners of the LAR receptor protein tyrosine phosphatase family, and to promote dimerization of Slitrk1. Altogether, our results demonstrate that Slitrk family proteins regulate synapse formation. One of the key steps in the development of the nervous system is the formation of new connections between different neurons. This process, referred to as synaptogenesis, also plays a critical role in the mature brain where the dynamic modification of circuitry has a profound effect on functions such as learning and memory. -

Rnai and Heterochromatin Repress Centromeric Meiotic Recombination
RNAi and heterochromatin repress centromeric meiotic recombination Chad Ellermeiera,1, Emily C. Higuchia, Naina Phadnisa, Laerke Holma,b, Jennifer L. Geelhooda, Genevieve Thonb, and Gerald R. Smitha,2 aDivision of Basic Sciences, Fred Hutchinson Cancer Research Center, Seattle, WA 98109; and bDepartment of Molecular Biology, University of Copenhagen Biocenter, DK-2200 Copenhagen, Denmark Edited* by Paul Nurse, The Rockefeller University, New York, NY, and approved April 2, 2010 (received for review December 9, 2009) During meiosis, the formation of viable haploid gametes from diploid correlated with birth defects resulting from chromosome mis- precursors requires that each homologous chromosome pair be segregation (2). (Here and subsequently, “centromeric” is meant to properly segregated toproduce anexact haploid set ofchromosomes. include “pericentromeric.”) Thus, repression of recombination spe- Genetic recombination, which provides a physical connection be- cifically in the centromere is crucial for the proper segregation of tween homologous chromosomes, is essential in most species for meiotic chromosomes, but the mechanism by which centromeric proper homologue segregation. Nevertheless, recombination is re- recombination is repressed during meiosis has been largely unknown. pressed specifically in and around the centromeres of chromosomes, Centromeric heterochromatin in many species represses apparently because rare centromeric (or pericentromeric) recombina- within its domain the abundance of transcripts and the expres- tion events, when they do occur, can disrupt proper segregation and sion of genes inserted into the heterochromatic region (4). In the lead to genetic disabilities, including birth defects. The basis by which fission yeast Schizosaccharomyces pombe, the formation of cen- centromeric meiotic recombination is repressed has been largely tromeric heterochromatin is facilitated by RNAi functions, which unknown. -

UBE4B Antibody (C-Term) Blocking Peptide Synthetic Peptide Catalog # Bp2111b
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 UBE4B Antibody (C-term) Blocking Peptide Synthetic peptide Catalog # BP2111b Specification UBE4B Antibody (C-term) Blocking Peptide UBE4B Antibody (C-term) Blocking Peptide - - Background Product Information Ubiquitin is a 76 amino acid highly conserved Primary Accession O95155 eukaryotic polypeptide that selectively marks cellular proteins for proteolytic degradation by the 26S proteasome. The process of target UBE4B Antibody (C-term) Blocking Peptide - Additional Information selection, covalent attachment and shuttle to the 26S proteasome is a vital means of regulating the concentrations of key regulatory Gene ID 10277 proteins in the cell by limiting their lifespans. Polyubiquitination is a common feature of this Other Names modification. Serial steps for modification Ubiquitin conjugation factor E4 B, 632-, include the activation of ubiquitin, an UBE4B (<a href="http://www.genenames.or ATP-dependent formation of a thioester bond g/cgi-bin/gene_symbol_report?hgnc_id=125 between ubiquitin and the enzyme E1, transfer 00" target="_blank">HGNC:12500</a>) by transacylation of ubiquitin from E1 to the Target/Specificity ubiquitin conjugating enzyme E2, and covalent The synthetic peptide sequence used to linkage to the target protein directly by E2 or generate the antibody <a href=/product/pr via E3 ligase enzyme. Deubiquitination oducts/AP2111b>AP2111b</a> was enzymes also exist to reverse the marking of selected from the C-term region of human protein substrates. Posttranslational tagging by UBE4B . A 10 to 100 fold molar excess to Ub is involved in a multitude of cellular antibody is recommended.