Examination and Processing of Human Semen
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
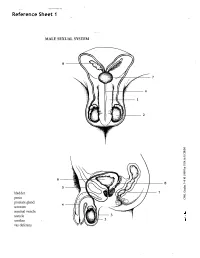
Reference Sheet 1
MALE SEXUAL SYSTEM 8 7 8 OJ 7 .£l"00\.....• ;:; ::>0\~ <Il '"~IQ)I"->. ~cru::>s ~ 6 5 bladder penis prostate gland 4 scrotum seminal vesicle testicle urethra vas deferens FEMALE SEXUAL SYSTEM 2 1 8 " \ 5 ... - ... j 4 labia \ ""\ bladderFallopian"k. "'"f"";".'''¥'&.tube\'WIT / I cervixt r r' \ \ clitorisurethrauterus 7 \ ~~ ;~f4f~ ~:iJ 3 ovaryvagina / ~ 2 / \ \\"- 9 6 adapted from F.L.A.S.H. Reproductive System Reference Sheet 3: GLOSSARY Anus – The opening in the buttocks from which bowel movements come when a person goes to the bathroom. It is part of the digestive system; it gets rid of body wastes. Buttocks – The medical word for a person’s “bottom” or “rear end.” Cervix – The opening of the uterus into the vagina. Circumcision – An operation to remove the foreskin from the penis. Cowper’s Glands – Glands on either side of the urethra that make a discharge which lines the urethra when a man gets an erection, making it less acid-like to protect the sperm. Clitoris – The part of the female genitals that’s full of nerves and becomes erect. It has a glans and a shaft like the penis, but only its glans is on the out side of the body, and it’s much smaller. Discharge – Liquid. Urine and semen are kinds of discharge, but the word is usually used to describe either the normal wetness of the vagina or the abnormal wetness that may come from an infection in the penis or vagina. Duct – Tube, the fallopian tubes may be called oviducts, because they are the path for an ovum. -

Glossary - Cellbiology
1 Glossary - Cellbiology Blotting: (Blot Analysis) Widely used biochemical technique for detecting the presence of specific macromolecules (proteins, mRNAs, or DNA sequences) in a mixture. A sample first is separated on an agarose or polyacrylamide gel usually under denaturing conditions; the separated components are transferred (blotting) to a nitrocellulose sheet, which is exposed to a radiolabeled molecule that specifically binds to the macromolecule of interest, and then subjected to autoradiography. Northern B.: mRNAs are detected with a complementary DNA; Southern B.: DNA restriction fragments are detected with complementary nucleotide sequences; Western B.: Proteins are detected by specific antibodies. Cell: The fundamental unit of living organisms. Cells are bounded by a lipid-containing plasma membrane, containing the central nucleus, and the cytoplasm. Cells are generally capable of independent reproduction. More complex cells like Eukaryotes have various compartments (organelles) where special tasks essential for the survival of the cell take place. Cytoplasm: Viscous contents of a cell that are contained within the plasma membrane but, in eukaryotic cells, outside the nucleus. The part of the cytoplasm not contained in any organelle is called the Cytosol. Cytoskeleton: (Gk. ) Three dimensional network of fibrous elements, allowing precisely regulated movements of cell parts, transport organelles, and help to maintain a cell’s shape. • Actin filament: (Microfilaments) Ubiquitous eukaryotic cytoskeletal proteins (one end is attached to the cell-cortex) of two “twisted“ actin monomers; are important in the structural support and movement of cells. Each actin filament (F-actin) consists of two strands of globular subunits (G-Actin) wrapped around each other to form a polarized unit (high ionic cytoplasm lead to the formation of AF, whereas low ion-concentration disassembles AF). -

MS Hatch-Waxman PTE REQUEST for EXTENSION of PATENT
Docket No.: 029420.0155-US02 (PATENT) IN THE UNITED STATES PATENT AND TRADEMARK OFFICE In re United States Patent of: Alan J. Korman, et aL. Patent No.: 7,605,238 Issued: October 20, 2009 For: HUMAN CTLA-4 ANTIBODIES MS Hatch-Waxman PTE Commissioner for Patents Offce of Patent Legal Administration Room MDW 7D55 600 Dulany Street (Madison Building) Alexandria, VA 223 14 REQUEST FOR EXTENSION OF PATENT TERM UNDER 35 U.S.C. §156 Sir: Pursuant to 35 U.S.C. §156 and 37 C.F.R. §§1.710-1.791, Medarex, Inc., the current address of which is Route 206 and Province Line Road, Princeton, New Jersey 08540 ("Applicant" or "Medarex"), hereby requests an extension of U.S. Patent No. 7,605,238 (the "'238 patent"). As permitted by 37 C.F.R. §1.785(b) and MPEP §2761, Applicant is concurrently fiing a request for patent term extension of U.S. Patent No. 6,984,720 based upon the same regulatory review period. Medarex represents that it is the owner and assignee of the entire interest in and to Letters Patent of the United States No. 7,605,238 (Exhibit 1) granted to Alan J. Korman, Edward L. Halk, Nils Lonberg, Yashwant M. Deo and Tibor P. Keler (the "inventors") on October 20, 2009, for "Human CTLA-4 Antibodies and Their Uses" by virtue of an assignment from the DC; 3983891.1 Patent No.: 7,605,238 - 2- Docket No.: 029420.0155-US02 Alan J. Korman, Edward L. Halk and Nils Lonberg to Medarex, recorded in the United States Patent and Trademark Offce ("PTO") on March 6, 2003 at Reel 013817, Frame 0628 and an assignment from Yashwant M. -

The Male Reproductive System
Management of Men’s Reproductive 3 Health Problems Men’s Reproductive Health Curriculum Management of Men’s Reproductive 3 Health Problems © 2003 EngenderHealth. All rights reserved. 440 Ninth Avenue New York, NY 10001 U.S.A. Telephone: 212-561-8000 Fax: 212-561-8067 e-mail: [email protected] www.engenderhealth.org This publication was made possible, in part, through support provided by the Office of Population, U.S. Agency for International Development (USAID), under the terms of cooperative agreement HRN-A-00-98-00042-00. The opinions expressed herein are those of the publisher and do not necessarily reflect the views of USAID. Cover design: Virginia Taddoni ISBN 1-885063-45-8 Printed in the United States of America. Printed on recycled paper. Library of Congress Cataloging-in-Publication Data Men’s reproductive health curriculum : management of men’s reproductive health problems. p. ; cm. Companion v. to: Introduction to men’s reproductive health services, and: Counseling and communicating with men. Includes bibliographical references. ISBN 1-885063-45-8 1. Andrology. 2. Human reproduction. 3. Generative organs, Male--Diseases--Treatment. I. EngenderHealth (Firm) II. Counseling and communicating with men. III. Title: Introduction to men’s reproductive health services. [DNLM: 1. Genital Diseases, Male. 2. Physical Examination--methods. 3. Reproductive Health Services. WJ 700 M5483 2003] QP253.M465 2003 616.6’5--dc22 2003063056 Contents Acknowledgments v Introduction vii 1 Disorders of the Male Reproductive System 1.1 The Male -

THE PHYSIOLOGY and ECOPHYSIOLOGY of EJACULATION Tropical and Subtropical Agroecosystems, Vol
Tropical and Subtropical Agroecosystems E-ISSN: 1870-0462 [email protected] Universidad Autónoma de Yucatán México Lucio, R. A.; Cruz, Y.; Pichardo, A. I.; Fuentes-Morales, M. R.; Fuentes-Farias, A.L.; Molina-Cerón, M. L.; Gutiérrez-Ospina, G. THE PHYSIOLOGY AND ECOPHYSIOLOGY OF EJACULATION Tropical and Subtropical Agroecosystems, vol. 15, núm. 1, 2012, pp. S113-S127 Universidad Autónoma de Yucatán Mérida, Yucatán, México Available in: http://www.redalyc.org/articulo.oa?id=93924484010 How to cite Complete issue Scientific Information System More information about this article Network of Scientific Journals from Latin America, the Caribbean, Spain and Portugal Journal's homepage in redalyc.org Non-profit academic project, developed under the open access initiative Tropical and Subtropical Agroecosystems, 15 (2012) SUP 1: S113 – S127 REVIEW [REVISIÓN] THE PHYSIOLOGY AND ECOPHYSIOLOGY OF EJACULATION [FISIOLOGÍA Y ECOFISIOLOGÍA DE LA EYACULACIÓN] R. A. Lucio1*, Y. Cruz1, A. I. Pichardo2, M. R. Fuentes-Morales1, A.L. Fuentes-Farias3, M. L. Molina-Cerón2 and G. Gutiérrez-Ospina2 1Centro Tlaxcala de Biología de la Conducta, Universidad Autónoma de Tlaxcala, Tlaxcala-Puebla km 1.5 s/n, Loma Xicotencatl, 90062, Tlaxcala, Tlax., México. 2Depto. Biología Celular y Fisiología, Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México, Ciudad Universitaria, 04510, México, D.F., México. 3Laboratorio de Ecofisiologia Animal, Departamento de Fisiologia, Instituto de Investigaciones sobre los Recursos Naturales, Universidad Michoacana de San Nicolás de Hidalgo, Av. San Juanito Itzicuaro s/n, Colonia Nueva Esperanza 58337, Morelia, Mich., México * Corresponding author ABSTRACT RESUMEN Different studies dealing with ejaculation view this Diferentes estudios enfocados en la eyaculación, process as a part of the male copulatory behavior. -

B2bi 2.5 Support Reference Catalog 3 Accessibility
B2Bi Version 2.5 April 2019 Support Reference Catalog Copyright © 2019 Axway All rights reserved. This documentation describes the following Axway software: Axway B2Bi 2.5 No part of this publication may be reproduced, transmitted, stored in a retrieval system, or translated into any human or computer language, in any form or by any means, electronic, mechanical, magnetic, optical, chemical, manual, or otherwise, without the prior written permission of the copyright owner, Axway. This document, provided for informational purposes only, may be subject to significant modification. The descriptions and information in this document may not necessarily accurately represent or reflect the current or planned functions of this product. Axway may change this publication, the product described herein, or both. These changes will be incorporated in new versions of this document. Axway does not warrant that this document is error free. Axway recognizes the rights of the holders of all trademarks used in its publications. The documentation may provide hyperlinks to third-party web sites or access to third-party content. Links and access to these sites are provided for your convenience only. Axway does not control, endorse or guarantee content found in such sites. Axway is not responsible for any content, associated links, resources or services associated with a third-party site. Axway shall not be liable for any loss or damage of any sort associated with your use of third-party content. Contents Accessibility 4 Accessibility features of B2Bi 4 Keyboard -

Sara Aghamohammadi, M.D
Sara Aghamohammadi, M.D. Philosophy of Care It is a privilege to care for children and their families during the time of their critical illness. I strive to incorporate the science and art of medicine in my everyday practice such that each child and family receives the best medical care in a supportive and respectful environment. Having grown up in the San Joaquin Valley, I am honored to join UC Davis Children's Hospital's team and contribute to the well-being of our community's children. Clinical Interests Dr. Aghamohammadi has always had a passion for education, she enjoys teaching principles of medicine, pediatrics, and critical care to medical students, residents, and nurses alike. Her clinical interests include standardization of practice in the PICU through the use of protocols. Her team has successfully implemented a sedation and analgesia protocol in the PICU, and she helped develop the high-flow nasal cannula protocol for bronchiolitis. Additionally, she has been involved in the development of pediatric pain order sets and is part of a multi-disciplinary team to address acute and chronic pain in pediatric patients. Research/Academic Interests Dr. Aghamohammadi has been passionate about Physician Health and Well-being and heads the Wellness Committee for the Department of Pediatrics. Additionally, she is a part of the Department Wellness Champions for the UC Davis Health System and has given presentations on the importance of Physician Wellness. After completing training in Physician Health and Well-being, she now serves as a mentor for the Train-the-Trainer Physician Health and Well-being Fellowship. -

Nutrition in Andrology, Gynaecology and Obstetrics
Appendix No. 2 to the procedure of development and periodical review of syllabuses Nutrition in Andrology, Gynaecology and Obstetrics 1. Imprint Faculty name: English Division Syllabus (field of study, level and educational profile, form of studies, Medicine, 1st level studies, practical profile, full time e.g., Public Health, 1st level studies, practical profile, full time): Academic year: 2019/2020 Nutrition in Andrology, Gynaecology and Module/subject name: Obstetrics Subject code (from the Pensum system): Educational units: Department of Social Medicine and Public Health Head of the unit/s: Dr hab. n. med. Aneta Nitsch - Osuch Study year (the year during which the 1st-6th respective subject is taught): Study semester (the semester during which the respective subject is Winter and Summer semesters taught): Module/subject type (basic, corresponding to the field of study, Optional optional): Teachers (names and surnames and Anna Jagielska, MD degrees of all academic teachers of Aleksandra Kozłowska, BSc respective subjects): ERASMUS YES/NO (Is the subject available for students under the YES ERASMUS programme?): A person responsible for the syllabus (a person to which all comments to Anna Jagielska, MD the syllabus should be reported) Number of ECTS credits: 2 Page 1 of 4 Appendix No. 2 to the procedure of development and periodical review of syllabuses 2. Educational goals and aims The aim of the course is to provide students with: 1. The principles of nutrition during adolescence, adulthood and eldery. 2. The relationship between nutrition and fertility, fetal status and communicable diseases in the adults life. 3. Basics of dietary advices for men and women in the reproductive years. -

1997 Asa Program.Pdf
Friday, February 21 12:00 NOON- 11:00 PM Executive Council Meeting (lunch and supper served) (Chesapeake Room NB) Saturday, February 22 8:00-9:40 AM Postgraduate Course (Constellation 3:00-5:00 PM Postgraduate Course (Constellation Ballroom A) Ballroom A) 9:40-1 0:00 AM Refreshment Break 6:00-7:00 PM Student Mixer (Maryland Suites-Balti 10:00-12:00 NOON Postgraduate Course (Constellation more Room) Ballroom A) 7:00-9:00 PM ASA Welcoming Reception (Atrium 12:00-1 :00 PM Lunch (on your own) Lobby) 7:00-9:00 PM Exhibits Open (Constellation Ball I :00-2:40 PM Postgraduate Course (Constellation Ballroom A) rooms E, F) 2:40-3:00 PM Refreshment Break 9:00- 1 I :00 PM Executive Council Meeting (Chesa peake Room NB) Sunday, February 23 7:45-8:00 AM Welcome and Opening Remarks 12:00-1 :30 PM Women in Andrology Luncheon (Ches (Constellation Ballroom A) apeake Room NB) 8:00-9:00 AM Serono Lecture: "Genetics of Prostate Business Meeting 12:00-12:30 Cancer" Patrick Walsh (Constellation Speaker and Lunch 12:30-1:30 Ballroom A) I :30-3:00 PM Symposium I: "Regulation of Testicu 9:00-10:00 AM American Urological Association Lec lar Growth and Function" (Constellation ture: "New Medical Treatments of Im Ballroom A) potence" Irwin Goldstein (Constella Patricia Morris tion Ballroom A) Martin Matzuk 10:00-10:30 AM Refreshment Break/Exhibits 3:00-3:30 PM Refreshment Break/Exhibits (Constellation Ballrooms E, F) (Constellation Ballrooms E, F) 10:30-12:00 NOON Oral Session I: "Genes and Male Repro 3:30-4:30 PM Oral Session II: "Calcium Channels duction" (Constellation Ballroom A) and Male Reproduction" (Constellation Ballroom A) 12:00- 1 :30 PM Lune (on your own) � 4:30-6:30 PM Poster Session I (Constellation Ball �4·< rooms C, D) \v\wr 7:30-11:00 PM Banquet (National Aquarium) Monday, February 24 7:00-8:00 AM Past Presidents' Breakfast 12:00-1 :30 PM Simultaneous Events: (Pratt/Calvert Rooms) I. -

Andrology User Handbook
Document code: AY.P001 Version number: 7 Date of issue: 29/06/2020 USER HANDBOOK FOR ANDROLOGY SERVICES Diagnostic Semen Analysis Post Vasectomy Semen Analysis Retrograde Ejaculation Analysis Page 1 of 24 Document code: AY.P001 Version number: 7 Date of issue: 29/06/2020 Contents: 1. Introduction .................................................................................................................... 3 2. Location and Opening Times .......................................................................................... 4 3. Useful contacts ............................................................................................................... 4 4. Services provided by the laboratory ............................................................................... 5 5. Requesting semen analysis ............................................................................................ 5 6. Analysis test types .......................................................................................................... 8 6.1 Diagnostic semen analysis (DSA) test for fertility ......................................................... 8 6.1a Instructions for collection of a semen sample for DSA (fertility)............................... 9 6.1b How Diagnostic Semen Analysis assessments are reported .................................10 6.2 Retrograde Analysis ....................................................................................................11 6.2a Instructions for collection of urine for retrograde ejaculation -

In 6861.Indd
Original Article Sexuality during gestation DOI: 10.5020/18061230.2018.6861 MALE PERCEPTION OF SEXUAL ACTIVITY IN THE GESTATIONAL PERIOD Percepção masculina sobre atividade sexual no período gestacional Percepción masculina sobre la actividad sexual en el período gestacional Dailon de Araújo Alves Regional University of Cariri (Universidade Regional do Cariri - URCA) - Crato (CE) - Brazil Brunna Suélli de Souza Alves Faculty of Juazeiro do Norte (Faculdade de Juazeiro do Norte - FJN) - Juazeiro do Norte (CE) - Brazil Willma José de Santana Faculty of Juazeiro do Norte (Faculdade de Juazeiro do Norte - FJN) - Juazeiro do Norte (CE) - Brazil Felice Teles Lira dos Santos Moreira Regional University of Cariri (Universidade Regional do Cariri - URCA) - Crato (CE) - Brazil Dayanne Rakelly de Oliveira Regional University of Cariri (Universidade Regional do Cariri - URCA) - Crato (CE) - Brazil Grayce Alencar Albuquerque Regional University of Cariri (Universidade Regional do Cariri - URCA) - Crato (CE) - Brazil ABSTRACT Objective: To describe men’s perception of the sexual activity during the gestational period, in the context of the daily life experienced with their pregnant partners. Methods: This is a descriptive study with a qualitative approach. The study included 10 spouses of pregnant women attended to at Basic Health Units in the city of Juazeiro do Norte, Ceará, Brazil. Data was collected between September and October 2015, through a semi-structured interview and evaluated through the systematic technique of content analysis, and analyzed in light of the pertinent literature. Results: The majority of interviewees belonged to the age group between 24 and 29 years, attended high school and were married. For the study participants, when it comes to sexuality, some understand it as something beyond sexual intercourse, whereas, for others, sexuality is related only to intercourse. -

Sexual Dysfunction and Related Factors in Pregnancy
Banaei et al. Systematic Reviews (2019) 8:161 https://doi.org/10.1186/s13643-019-1079-4 PROTOCOL Open Access Sexual dysfunction and related factors in pregnancy and postpartum: a systematic review and meta-analysis protocol Mojdeh Banaei1, Maryam Azizi2, Azam Moridi3, Sareh Dashti4, Asiyeh Pormehr Yabandeh3 and Nasibeh Roozbeh3* Abstract Background: Sexual dysfunction refers to a chain of psychiatric, individual, and couple’s experiences that manifests itself as a dysfunction in sexual desire, sexual arousal, orgasm, and pain during intercourse. The aim of this systematic review will be to assess the sexual dysfunction and determine the relevant factors to sexual dysfunction during pregnancy and postpartum. Methods and analysis: All observational studies, including descriptive, descriptive-analytic, case-control, and cohort studies published between 1990 and 2019, will be included in the study. Review articles, case studies, case reports, letter to editors, pilot studies, and editorial will be excluded from the study. The search will be conducted in the Cochrane Central Register, MEDLINE, Google Scholar, EMBASE, ProQuest, Scopus, WOS, and CINAHL databases. Eligible studies should assess at least one of the sexual dysfunction symptoms in pregnant women orinthefirstyearpostpartum.Quality assessment of studies will be performed by two authors independently based on the NOS checklist. This checklist is designed to assess the quality of observational studies. Data will be analyzed using Stata software ver. 11. Considering that the index investigated in the present study will be the level of sexual disorder, standard error will be calculated for each study using binomial distribution. The heterogeneity level will be investigated using Cochran’sQstatisticandI2 index in a chi-square test at a significance level of 1.1.