Selective Irradiation NMR Experiments: Homonuclear Decoupling and the NOE Difference Experiment
OBJECTIVE: To demonstrate proficiency in obtaining (1) a spectrum using homonuclear decoupling to assign J-coupled protons, and (2) a 1D-NOE difference spectrum to detect spatially proximal protons. The compound under study is a half-cage isodrin derivative employed in the original demonstration of the nuclear Overhauser effect. You will also learn how to prepare a degassed (oxygen-free) sample for NOE measurements, using the “freeze-pump-thaw” method on a vacuum manifold. After a year of introductory organic chemistry, most students view NMR as a spectroscopic tool useful for assigning bond connectivity on the basis of chemical shifts and scalar (through-bond) coupling patterns. A third important observeable, the NOE, arises as a consequence of through-space interactions among nuclei possessing spin. This laboratory will equip you to identify scalar-coupled protons by a simple decoupling experiment and also to identify spatially proximal protons via a 1D NOE difference measurement. These two experiments are among the most heavily used one-dimensional proton experiments by practitioners of NMR. It is highly likely that a first- or second-year graduate student in organic chemistry would be expected to know how to execute either of these two experiments in conjunction with any research problems requiring structure elucidation.
PRE-LAB DISCUSSION: Read this material and prepare a short presentation on the two pulse programs (zghd and zgf2pr) used for this project. How do they work?
PART 1: THE HOMONUCLEAR DECOUPLING EXPERIMENT
BACKGROUND
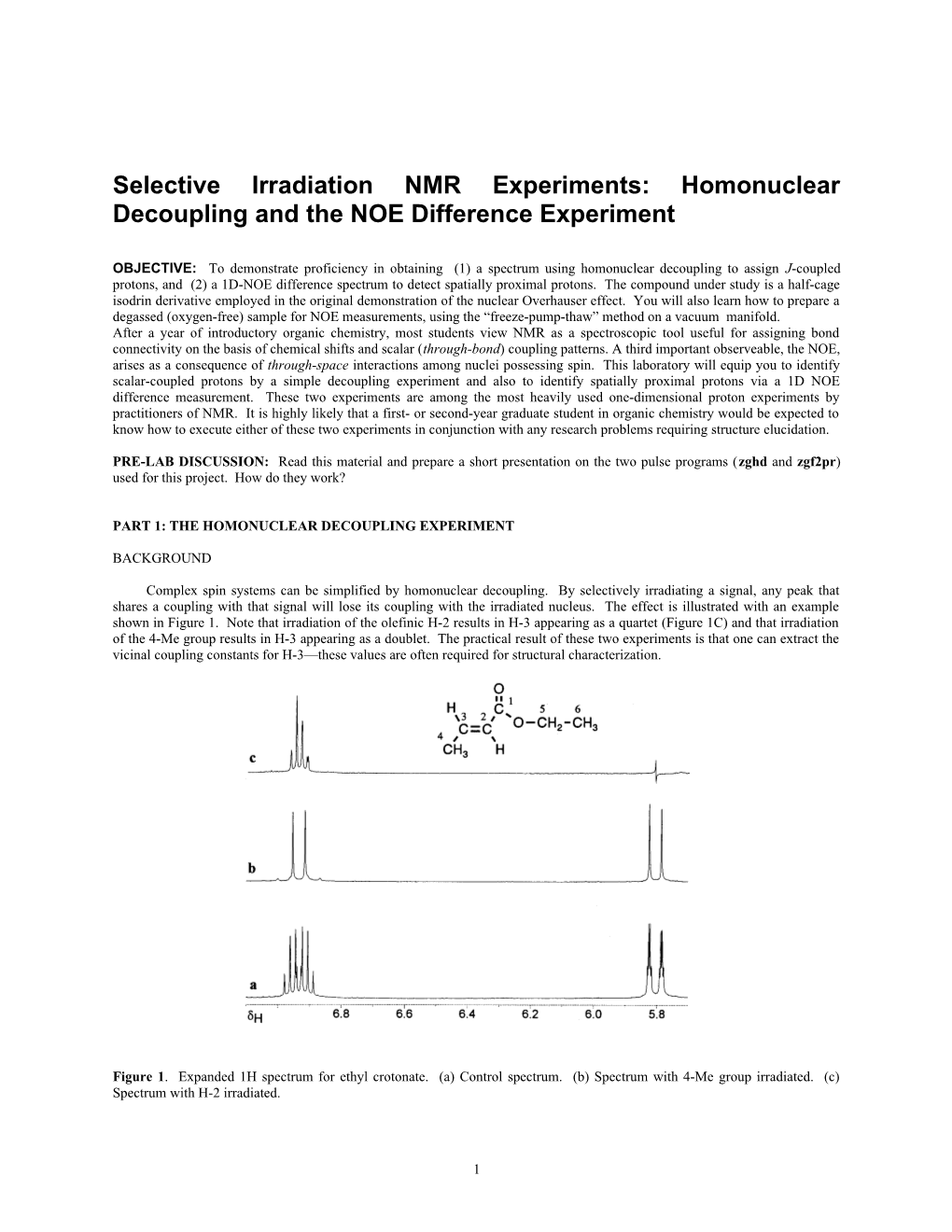
Complex spin systems can be simplified by homonuclear decoupling. By selectively irradiating a signal, any peak that shares a coupling with that signal will lose its coupling with the irradiated nucleus. The effect is illustrated with an example shown in Figure 1. Note that irradiation of the olefinic H-2 results in H-3 appearing as a quartet (Figure 1C) and that irradiation of the 4-Me group results in H-3 appearing as a doublet. The practical result of these two experiments is that one can extract the vicinal coupling constants for H-3—these values are often required for structural characterization.
Figure 1. Expanded 1H spectrum for ethyl crotonate. (a) Control spectrum. (b) Spectrum with 4-Me group irradiated. (c) Spectrum with H-2 irradiated.
1 Why does the signal for H-2 disappear while it is being irradiated, as shown in Figure 1c? To answer this question, we must consider the pulse program used to acquire a spectrum with homonuclear decoupling, and how that pulse program affects the nuclear spin states. A more simple two-spin system is considered in Figure 2. The case of two spins (H a and Hx) with different chemical shifts and no spin-spin coupling is shown in Figure 1a. A four-level energy diagram is written to demonstrate that such a system would exhibit different chemical shifts for Ha and Hb. Note that the energy spacing between single-quantum transitions for a given spin is degenerate (e.g., 1-3 and 2-4 for H a) yet different from those of the other spin (Figure 1b). If a coupling mechanism is operative (scalar coupling is nuclear spin information transmitted from one nucleus to another via electrons residing in (usually) covalent bonds connecting the nuclei), then the energy levels are perturbed such that the single quantum transitions for a given nucleus are no longer degenerate (note that the 1-3 and 2-4 transitions for H a are no longer equivalent). The action of a decoupling field placed at the Hx chemical shift (Figure 2c) serves to remove the identity of the
Hx spins, and the result is that the signal for the Ha spins collapses to a singlet at the Ha chemical shift.
c b a
Hx irradiated J > 0 J = 0 4 3
2 1 Irradiate 2-4 Jax Jax 2-4 3-4 1-3 2-4 1-3 3-4 1-2 1-3 1-2
H Hx H H a Ha x Ha x
Descriptors: =
Ha()Hx()
Figure 2. (a) Energy level diagram for two spins Ha and Hx. Alignment of nuclear magnetization with respect to spectrometer magnetic field defines relative energies (=with field, =against field). Note that when J=0, two transitions contribute to each signal (Ha signal is comprised of single quantum transitions from levels 1-3 and 2-4, respectively. (b) For spins sharing a scalar coupling, the energy levels are perturbed by the coupling mechanism. Note that the transition energies are now different and give rise to a splitting (Jax) for each peak. (c) Under the action of a decoupler field positioned at the H x chemical shift, the Hx spin states ( vs ) are no longer defined, and the Ha transition frequency collapses to the unperturbed value, i.e., Ha appears as a singlet.
The pulse program used in a homonuclear decoupling experiment is illustrated in Figure 3. A frequency-selective irradiation source is applied during the data acquisition period (AQ), and the only “trick” to getting this experiment to work correctly is making sure that the decoupler is configured to irradiate with the appropriate power level at the correct frequency.
2 1H decoupler power pl24
1H channel
d1 p1 aq
Figure 3. Pulse sequence for homonuclear decoupling sequence.
SETTING UP THE EXPERIMENT
1. We will use the same sample for the decoupling experiment and the NOE difference measurement. You will prepare a
15 mg/ml sample of half-cage propionate 4 dissolved in acetone-d6 in a J-Young NMR tube. Your instructor will show you how to remove dissolved gases in the sample using the freeze-pump-thaw method. Place the sample in the magnet, tune and shim, and then obtain a properly phased 1H spectrum of the sample (open file dec_setup, create your own copy of this file with the “new” command). Resident pulse program: zg 2. Turn on the f2 channel with the following steps. Type eda. Select NUCLEI edit. In the submenu click on the box labeled “off” under the f2 button and define 1H as the irradiation frequency. Select SAVE to return to the eda menu, then select SAVE again. Use this spectrum to set the O2 frequency for f2 decoupling. Enter the utilities subroutine. Select the button labeled O2. This will put the display into the mode where the arrow is “stuck” to the spectrum. Position the arrow on the resonance that you want to irradiate. It is suggested that you irradiate the high field triplet. Press the middle button to define the frequency. This will set the value of the O2 parameter. Select return to exit the utilities subroutine. 3. If you do not want to overwrite your original spectrum, create a new data set (New, change the experiment number). 4. Select a pulse program that will control the timing and power of the 1H irradiation. Type eda, select the PULPROG parameter (top left). Type in the name zghd. Set the parameter DIGMOD (lower right) to homodecoupling-digital. Select SAVE to exit the eda menu. Set pl24 to 65 dB. Pl24 (the letters p and l) is the decoupler power. Its units are decibels; higher values mean lower power. Set d12 to 20 microseconds (enter 20 terminate with the letter u). Record a spectrum and compare the appearance of the ethyl resonance with that you observed in the undecoupled spectrum. Record several additional spectra (change the expt number each time with the New command) and “map out” the values of pl24 (changing in increments of 5 dB) required to (i) weakly perturb the CH2 resonance, (ii) cleanly collapse the CH2 resonance to a singlet. 5. Record plots of your work and describe your findings.
CALIBRATING THE DECOUPLER ATTENUATION
1. Read pages 90-92 of Berger’s book (included in the appendix of this handout) and execute a series of experiments to calibrate the decoupler bandwith as a function of decoupler attenuation. Develop a calibration curve as shown on page 92. Compare your calibration curve with the empirical results obtained in the first section.
3 PART 2: THE NOE DIFFERENCE EXPERIMENT
BACKGROUND
The Nuclear Overhauser Effect (NOE), a method for probing through-space interactions between nuclei, has emerged as one of the most useful experiments in NMR spectroscopy. The original electron-nuclear experiment, conceived by Overhauser (1,2) and implemented by Carver and Slichter (3,4), measured NMR signal enhancements in lithium metal while saturating the electron spins. A similar effect arising from the interaction of two nuclei was demonstrated by Solomon (5) in a double resonance study of liquid hydrogen fluoride, HF. The utility of NOE experiments in structural studies of organic compounds was first reported in 1965 by Anet and Bourn (6). Three molecules were used in that study: ,-dimethylacrylic acid (1), N,N-dimethyl- formamide (2), and “half-cage” acetate 3, a compound with the shortest known H-H distance in an organic compound. The technique soon found application in structural studies of natural products, e.g., taxane derivatives, in the pioneering work by Nakanishi and co-workers (7,8). In 1972, Bothner-By and co-workers (9) described negative nuclear Overhauser effects in the interaction of small molecules with proteins, and the advent of Ernst’s two-dimensional NOESY experiment (10) ushered in a new era, particularly in macromolecular structure elucidation. A tremendous number of applications of the Overhauser effect have appeared and are summarized in two texts (11,12).
C l H H O R H C C O O H H C O C l 3 3 9 5 N C l C l O H 3 C H H 3 C H 3 R = C H C l 3 1 2 C l 4 R = C H 2 C H 3
Half-cage 4 contains two protons (H-5 and H-9) separated by an ultrashort nonbonded H-H distance of 1.62 Å as illustrated in figure 4 (13). It is unusual to have protons situated so close to each other and not be scalar coupled to each other (14). Large Overhauser effects are expected for pairs of nuclei that are isolated from other magnetic nuclei, and the H-5/H-9 pair is reasonably isolated (the distances of other protons to H-5 and H-9 are all larger than 2.7 Å). This means that these distant protons essentially can be ignored in considering the relaxation of the H-5,H-9 pair and the NOE within that pair. This isolation from the other nuclei gives rise to an exceptionally large NOE (ca. +43%). The effect is easy to observe and quantify because the relevant (T1) relaxation times are short, which is a consequence of the close proximity of H-5 and H-9 and the medium size of the molecule.
Figure 4. Half-Cage Benzoate Structure (the Me or Et in 3 or 4 has been replaced by a Phenyl) as Determined by Neutron Diffraction Analysis (Ref. 13).
The NOE is a manifestation of NMR relaxation pathways among adjacent nuclei possessing magnetic dipoles. Spin-lattice (T1) relaxation is the process by which nuclear spins attain their equilibrium (Boltzmann) populations within the spectrometer magnetic field. 1/T1 describes the rate at which the sample becomes polarized when placed in the magnet or after the application of a pulse, which might have perturbed the equilibrium polarization (e.g., a 90° pulse to generate a spectrum). Spectroscopists usually talk about quantized states, and NMR relaxation describes the time it takes for quantized spin states to develop. Because the spin states are quantized, the energy required to induce transitions (a requirement for relaxation) must satisfy the usual requirement (E = h).
Dipole-dipole relaxation is the source of the Overhauser effect and one considers the spin-lattice relaxation (T1) behavior to identify how the NOE changes with relative internuclear distance, competitive relaxation, and molecular tumbling. Nuclei possessing spin act as small magnetic dipoles that generate relatively strong local magnetic fields. For example, the magnetic field 1.6 Å away from a proton is 0.4 mT, which is far less than the spectrometer’s static magnetic field (9.4 T for 1H at 400 MHz). However, 0.4 mT is much greater than the typical 1H chemical shift range (10 ppm at 9.4 T is 94 T) or for that matter, the earth’s magnetic field (ca. 50 T). Due to rapid motional averaging, these strong local dipolar fields do not affect the line positions or intensities in NMR spectra of small molecules. Instead, dipolar fields give rise to relaxation effects.
4 2 I S
1 Q 1 Q H H I S 4 4 I S I S I S I S I S I S 1 Q 1 Q I S I S I S I S
I r r a d i a t e a' a 6 I S I n t e n s i t y : 4 4
3 I S
5 I S I S 3 I r r a d i a t e b'
I n t e n s i t y : 4
b 5 I S
I r r a d i a t e 2 . 5 I S c'
I n t e n s i t y : 6 2 Q
0 Q I S 5 . 5 I S 2 . 5 d
I n t e n s i t y : 2 - 4 c 5 . 5 I S
Figure 5. (a) Energy level diagram for 16 dipolar-coupled spin pairs (HI and HS), at equilibrium, aligned with () or against () the spectrometer magnetic field. Single quantum transitions are labeled as 1Q and levels are populated with arbitrary numbers to illustrate the Boltzmann distributions and signal intensities. (a’) Hypothetical NMR spectrum resulting from a nonselective pulse of the spin system described in 5a, “off resonance” irradiation does not affect the energy levels or the observed spectrum. (b) The effect of selectively irradiating 1Q transitions (wavy lines) associated with spins HS: populations across these transitions are immediately equalized. (b’) Hypothetical NMR spectrum resulting from a nonselective pulse of the spin system described in 5b. Note that the signal for H S disappears as a consequence of the irradiation. (c) Irradiation as in 5b, only with sufficient time for dipolar relaxation to occur; for small molecules double quantum (2Q) transitions cause non-equilibrium population differences to develop. Namely, some of the population of the high-energy spin state is transferred to the low-energy spin state as a consequence of dipolar relaxation. The selective irradiation serves to continually equalize the populations as shown. Zero-quantum (0Q) transitions, shown with dotted line, do not appreciably contribute to relaxation. (c’) Hypothetical 1H spectrum resulting from a nonselective pulse of the spin system described in part 5c. (d) NOE difference spectrum resulting from the subtraction of spectrum a’ from spectrum c’.
5 To cause relaxation, components of the dipolar field must oscillate with frequency components matched to the appropriate nuclear spin transitions (Figure 5). The strength of the spectrometer magnetic field determines the transition frequencies: on a 9.4-Tesla instrument the single quantum (1Q) transition for a proton is 400 MHz. The spectrometer can induce such transitions with an RF pulse that creates an oscillating magnetic field in the sample; likewise, the spin pair will experience transitions (relaxation) caused by tumbling-induced oscillating magnetic fields. In a sense, the tumbling serves as the oscillator, with the tumbling rate determined by the size and shape of the molecule, the viscosity of the medium, and the temperature. The equation 15 for T1 relaxation for intramolecular dipolar relaxation between two spins is given by ( ):
1 3 1 4 ( 2 R ) 2 c 2 2 2 2 . (1) T 1 1 0 1 c 1 4 c
3 In Equation 1 R is the “dipolar coupling constant”, in Hz, and is defined as (0/4)12( h /2)r , where 0 is the -7 -2 -2 7 -1 -1 permeability constant (4 10 kg m s A ). 1 and 2 are the respective proton magnetogyric ratios (H = 26.752 10 T s ), h is the Planck constant (1.05 10-34 J s), and r is the internuclear distance (1Å = 1 10-10 m). The remaining terms in Equation -1 6 -1 1 are the Larmor frequency (expressed in s , 300 MHz x 2 = 1,885 10 s ) and the correlation time, c (expressed in seconds), characterizing random molecular reorientation. Equation 1 assumes isotropic molecular motion, which means the random reorientation rate is the same for all axes in the molecule (a sphere tumbles isotropically). Rotational diffusion in the 16 half-cage compounds has been shown to be roughly isotropic ( ). Figure 6 shows a graph of T1 vs. c to illustrate the behavior of Equation 1 for two protons situated 1.62 Å apart at spectrometer frequencies of 400 and 800 MHz.
1 0 0
8 0 0 M H z e x t r e m e n a r r o w i n g T ( s ) 1 0 r e g i o n 1
1 T m i n 1
4 0 0 M H z
0 . 1 - 1 2 - 1 1 - 1 0 - 9 - 8 l o g ( s ) c
Figure 6. T1 vs. tc for intramolecular dipole-dipole relaxation for two protons separated by 1.62 Å at spectrometer frequencies of 400 MHz (solid line) and 800 MHz (dashed line) using Equation 1 as parameterized in the text.
With increasing molecular size, T1 first decreases, passes through a minimum, and then becomes longer. The region in which molecular tumbling is rapid (<<1) is termed the extreme narrowing region, and it is characteristic of most small organic molecules at room temperature in solutions of reasonably low viscosity. In this region T1 is independent of the spectrometer frequency for a given sample and temperature. The graphical illustration of Equation 1 illustrates an important point: NMR dipolar relaxation phenomena, including the NOE, involve molecular motion and can be dependent on the strength of the spectrometer’s magnetic field.
The Overhauser enhancement is related to the resonance frequency and the tumbling time as shown in Equation 2:
5 2 2 4 4 4 c c (2) 2 2 4 4 1 0 2 3 c 4 c
Equation 2 is a condensed form of a more complex equation relating molecular motion to all of the possible transition or “spin flip” frequencies (Figure 5). For a two spin system (H-I and H-S) one needs to consider:
single quantum transitions (1Q) occurring at the Larmor frequency I or S. These are the transitions one observes in an NMR spectrum.
a double quantum transition (2Q) occurring at a higher frequency I +S or when two spins aligned with the field mutually flip to an orientation against the field or vice-versa. As discussed earlier, it is this transition that gives rise to the positive NOE in small and medium-sized molecules (Figure 2).
a zero quantum transition at a lower frequency I —S when two opposed spins exchange orientation with respect to the field. This low-frequency transition is responsible for negative NOEs in slowly tumbling macromolecules.
6 For small molecules in the extreme narrowing region (<<1), Equation 2 predicts the maximum homonuclear (e.g., 1H-1H) enhancement is 50%, or = 5/10.
0 . 5
N O E 0 4 0 0 M H z
8 0 0 M H z
- 0 . 5
- 1 0 . 0 1 0 . 1 1 1 0 1 0 0 c
Figure 7. The maximum homonuclear NOE () as a function of c using Equation 2 with spectrometer frequencies of 400 and 800 MHz.
A plot of Equation 2 (Figure 7) serves to illustrate that the magnitude and sign of the nuclear Overhauser effect varies as a function of the spectrometer frequency and molecular tumbling. Using the T1 value (0.56 s at 400 MHz) for H-9 in 4 (chosen as the proton with the least amount of competitive relaxation -11 with other protons), one can estimate c using Equation 1 and the H-9,H-5 internuclear distance: c = 3.9 10 s, thus c = 0.098 and = 0.49. This analysis reveals that half-cage 4 is not quite completely in the extreme narrowing motional regime and is therefore subject to small field-dependent relaxation effects. At 800 MHz, the highest proton resonance frequency that is currently commercially available (at the time of this writing, a 900 MHz instrument has been installed at the Scripps Research Institute), the same c gives c = 0.196 and = 0.46, or in other words the maximum NOE is diminished at higher resonance frequencies. The above analysis reveals that for smaller organic molecules, there are certain benefits obtained by working at lower magnetic field strengths! It is also apparent from Figure 7 that under some circumstances (governed by a combination of molecular tumbling rate and spectrometer frequency) the nuclear Overhauser effect can be zero or nearly zero. This complication can be overcome by using rotating frame experiments (12). The large and negative enhancements, typical of slowly tumbling macromolecules, have provided a rich source of through-space information in structural studies of biologically relevant molecules. The +43% NOE in half-cage 4 should be regarded as a special case when compared to smaller yet structurally useful enhancements encountered in the research setting, which are typically 1-10% (17). One may ask why the NOE in 4 is not larger, as theory predicts a value of 49%. A complete analysis incorporating anisotropic tumbling and all of the proton-proton interactions revealed that the NOE at H-9 would be at best 47-48% ( 18). The origin of the ca. 4-5% discrepancy between experiment and theory remains elusive and is a topic of ongoing investigation.
SETTING UP THE EXPERIMENT
A 400 MHz 1H NMR spectrum of 4 is shown in Figure 8a. The peaks of interest, H-5 and H-9, can be assigned on the basis of downfield shifts caused by the electronegative Cl and OC(O)Et substituents and by steric compression ( 19). H-5 is assigned to the low-field resonance on the basis of its broad lineshape due to unresolved spin-spin couplings to adjacent hydrogen nuclei. Prior to embarking on NOE studies, it is advisable to measure the spin-lattice (T1) relaxation times; this information is useful for parameter selection in the NOE experiment. These values have already been determined for the sample you will be 20 using for this experiment. Using an inversion-recovery sequence, apparent T1 values ( ) of 0.56 s were obtained for H-5 and H-9 at 400 MHz.
7 c
H - 5 H - 9 b
a
1 Figure 8. (a) 400 MHz H spectrum of 4 dissolved in CDCl3. Each spectrum (16K points, spectral width = 8000 Hz, spectrometer frequency 400 MHz) consisted of 32 scans and was acquired with a 90° pulse following a 6.0 s irradiation period. A 6.0 s recycle delay was used between scans. (b) Expansion of H-5 and H-9 signals, control spectrum for NOE difference measurement. (c) Same region as in (b), the normal spectrum shown in (b) has been subtracted from a spectrum obtained after the resonance of H-5 had been saturated. The H-9 peak is now larger than in (b) because of the NOE, which in this case is a 43% enhancement of the intensity (the peak of H-5 is now negative because H-5 has zero intensity after saturation). Total experiment time: 20 minutes.
One measures the NOE (Figure 8) by irradiating a peak for a period of time long enough for nearby protons to become enhanced. The choice of irradiation time is rather arbitrary and system-dependent. In the study described here, H-5 was 21 irradiated for a period equal to 10 T1 or 6 s ( ). Overhauser effects for small to medium-sized molecules are observed as non- equilibrium, larger-than-normal signal intensities. The enhancement () is calculated by integration of a difference spectrum obtained by subtracting a control spectrum from one obtained with signal irradiation. One first integrates the irradiated signal and sets this value to —100%; the integrated intensity of any Overhauser-derived peak is then expressed as a percentage. As shown in Figures 8b and 8c, irradiation of H-5 gives a 43% enhancement at H-9. Question: can you sketch the pulse sequence used in the NOE difference measurement?
1. Obtain a properly phased 1H spectrum of the sample (open file noe_setup, create your own copy of this file with the “new” command). Resident pulse program: zg. Optimal NOE results are obtained with a NON-SPINNING sample. 2. Turn on the f2 channel with the following steps. Type eda. Select NUCLEI edit. In the submenu click on the box labeled “off” under the f2 button and define 1H as the irradiation frequency. Select SAVE to return to the eda menu, then select SAVE again. Use this spectrum to set the O2 frequency for f2 decoupling. Enter the utilities subroutine. Select the button labeled O2. This will put the display into the mode where the arrow is “stuck” to the spectrum. Position the arrow on the resonance that you want to irradiate. It is suggested that you irradiate the H-5 resonance. Press the middle button to define the frequency. This will set the value of the O2 parameter. Select return to exit the utilities subroutine. If you do not want to overwrite your original spectrum, create a new data set (New, change the experiment number). 3. Select a pulse program that will control the timing and power of the 1H irradiation. Type eda, select the PULPROG parameter (top left). Type in the name zgf2pr. Set pl14 to 70 dB. Pl14 (the letters p and l, the number 14) is the decoupler power. Its units are decibels; higher values mean lower power. Change d1 to 5 sec. This is the delay before the pulse during which the irradiation occurs. Set d11 to 30 msec and d13 to 3 microseconds. Record an 8 scan spectrum and inspect the H-5 resonance. If your decoupler power and irradiation time are correctly set, there should be very little signal intensity remaining. If there is, then adjust the decoupler power pl14 until the peak is saturated. This spectrum will serve as the on-resonance irradiation—you now need to collect the off-resonance control spectrum. Before proceeding to step 4, define O2 to be ca. 200 Hz downfield of the H-5 resonance (use the procedure outlined in step 2). 4. Change the experiment number with the new command. Record a spectrum and phase it using the same phase correction as used for the on-resonance irradiation spectrum: ft, pk (pk should apply the phase constants stored from the prior spectrum. 5. From the job containing your on-resonance irradiation, go into dual display mode by typing dual, define the second data set as your off-resonance irradiation data set; be sure to set the procno for the second data set to “1” (the default is “2” for some reason). In dual display mode, the second data set is purple and the “resident” data set is green. Subtract the off- resonance spectrum from the on-resonance spectrum by depressing the DIFF button at left. Save the subtraction as a new
8 data set by pressing return, then save and return: the result will be saved to the current filename and expno (experiment number) but to a different procno (the processed data file = 2). You can read in the original on-resonance spectrum with the command re expno procno (e.g., re 4 1 [re 4 space 1] will return the original data while re 4 2 will return the difference spectrum). Integrate the data and determine the percentage enhancement. 6. Record plots of your work and describe your findings.
The process described above is for a single irradiation employing a limited number of scans. In the general case, you might have to signal average for 256-512 scans in order to detect very small NOEs using the difference technique. In this case, it is best to collect the on-resonance/off-resonance data in an interleaved mode. This is preferable to obtaining 256 scans on-resonance and then 256 scans off resonance because the conditions of data acquisition (temperature, rf components, etc.) may change over the duration of a long acquisition. A procedure such as this is possible, though a different method for data acquisition is employed:
1. Obtain a properly phased 1H spectrum of the sample (open file noe_setup, create your own copy of this file with the “new” command). Optimal NOE results are obtained with a NON-SPINNING sample. 2. Turn on the f2 channel with the following steps. Type eda. Select NUCLEI edit. In the submenu click on the box labeled “off” under the f2 button and define 1H as the irradiation frequency. Select SAVE to return to the eda menu, then select SAVE again. Use this spectrum to set the O2 frequency for f2 decoupling. Enter the utilities subroutine. Select the button labeled O2. Select the button labeled frqlist. The following dialog will occur: a. Please enter type of list: f1 b. Please enter name of f1 list: input desired list name (e.g., frqlist_160) c. Write name of f2 list to acqu parameters? y d. (If the list exists, will get the following: Freq. list exists, append (a), overwrite (o), or quit (q) e. At this point, you will use the mouse to select the positions of the various irradiations. The message window will tell you of the three mouse button actions. Position the arrow on the resonance you want to irradiate, press the middle mouse button to define the frequency. Repeat for each irradiation point, and be sure to include a position on the list for the off-resonance irradiation. Processing will be easier if you make the off- resonance value either the first or the last on the list. When all irradiation points have been defined, press the left mouse button to terminate the selection process. At this point you have a frequency list whose name has been entered as the parameter f1list. It is suggested you irradiate at least 3 frequencies (one must be off- resonance). 3. Select return to exit the utilities subroutine. 4. If you do not want to overwrite your original spectrum, create a new data set (New, change the experiment number). 5. Select a pulse program that will control the timing and power of the 1H irradiation. Type eda, select the PULPROG parameter (top left). Type in the name zgf2pr. Set pl14 to the value you determined to be optimal from the prior section. Change d1 to 5 sec. This is the delay before the pulse during which the irradiation occurs. Set d11 to 30 msec and d13 to 3 microseconds. 6. Run the automation sequence noediff: type xau noediff. The data for each irradiation will be saved to sequential experiment numbers resident within the filename you have already selected. The following dialog will appear: f. Name of frequency list: frqlist_160 g. # of irradiation points: 3 h. # of time average cycles: 4 (this value controls how many times you will interleave datasets of 8 scans each). 7. Data is processed as before. 8. Record plots of your work and describe your findings.
9 ;zghd ; avance-version ;1D sequence with homodecoupling
#include
;;"d12=20u"
1 ze d12 pl24:f2 2 d1 p1 ph1 go=2 ph31 hd:f2 wr #0 d12 do:f2 exit ph1=0 2 2 0 1 3 3 1 ph31=0 2 2 0 1 3 3 1
;pl1 : f1 channel - power level for pulse (default) ;pl24: f2 channel - power level for hd/hc decoupling ;p1 : f1 channel - high power pulse ;d1 : relaxation delay; 1-5 * T1 ;d12: delay for power switching [20 usec]
;use DIGMOD homodecoupling-digital
10 ;zgf2pr ;avance-version ;1D sequence with f2 presaturation
#include
;;"d11=30m" ;;"d13=3u"
1 ze d11 pl14:f2 2 d1 cw:f2 d13 do:f2 p1 ph1 go=2 ph31 wr #0 exit ph1=0 2 2 0 1 3 3 1 ph31=0 2 2 0 1 3 3 1
;pl1 : f1 channel - power level for pulse (default) ;pl14: f2 channel - power level for cw decoupling ;p1 : f1 channel - 90 degree high power pulse ;d1 : relaxation delay; 1-5 * T1 ;d11: delay for disk I/O [30 msec] ;d13: short delay [3 usec]
11 (From Braun, Kalinowski, and Berger’s “150 and More Basic NMR Experiments: A Practical Course”)
12 13 14 Literature Cited
15 1. Overhauser, A.W. Phys. Rev. 1953, 91, 476. 2. Overhauser, A.W. Phys. Rev. 1953, 92, 411. 3. Carver, T.R.; Slichter, C.P. Phys. Rev. 1953, 92, 212. 4. Carver, T.R.; Slichter, C.P. Phys. Rev. 1956, 102, 975. 5. Solomon, I. Phys. Rev. 1955, 99, 559. 6. Anet, F. A. L.; Bourn, A. J. R. J. Am. Chem. Soc. 1965, 87, 5250. 7. Woods, M. C.; Miura, I.; Nakadaira, Y.; Terahara, A.; Maruyama, M.; Nakanishi, K. Tetrahedron Letters 1967, 321. 8. Woods, M. C.; Chiang, H.-C.; Nakadaira, Y.; Nakanishi, K. J. Am. Chem. Soc. 1968, 90, 522. 9. Balaram, P.; Bothner-By, A. A.; Dadok, J. J. Am. Chem. Soc. 1972, 94, 4015. 10 . Macura, S.; Ernst, R. R.; Mol. Phys. 1980, 41, 95. Kumar, A,; Ernst, R. R.; Wuthrich, K. Biochem. Biophys. Res. Comm. 1980, 95, 1. 11. Noggle, J. H.; Schirmer, R. E. “The Nuclear Overhauser Effect.” Academic Press, New York, 1971. 12 . Neuhas, D.; Williamson, M. P. “The Nuclear Overhauser Effect in Structural and Conformational Analysis.” VCH Publishers, New York, 1989. 13. Ermer, O.; Mason, S. A.; Anet, F. A. L.; Miura, S. S. J. Am. Chem. Soc. 1985, 107, 2330. 14 . Scalar coupling can complicate the analysis of NOE spectra, and many textbooks present NOE theory with the caveat that scalar coupling be ignored. 15. Harris, R. K. “Nuclear Magnetic Resonance Spectroscopy.” Longman Scientific & Technical, Essex, 1986. 16. Anet, F. A. L.; Dekmezian, A. Bull. Magn. Reson. 1980, 2, 6. 17 . Enhancements of less than 0.1% can now be reliably detected using a scheme recently described, see: Stonehouse, J.; Adell, P.; Keeler, J.; Shaka, A.J. J. Am. Chem. Soc. 1994, 116, 6037. 18. Miura, S. Ph. D. Thesis, University of California, Los Angeles, 1983. 19. Winstein, S.; Carter, P.; Anet, F.A.L.; Bourn, A.J.R. J. Am. Chem. Soc. 1965, 87, 5247.
20. The T1s were measured using an inversion-recovery sequence. 21 . The usual prescription is to irradiate for a period five times the T1 of the nucleus showing the enhancement. Despite its prevalence in the NMR literature, this particular duration does not appear to have any theoretical justification.