Remyelination in Multiple Sclerosis – How Close Are
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Oligodendrocytes in Development, Myelin Generation and Beyond
cells Review Oligodendrocytes in Development, Myelin Generation and Beyond Sarah Kuhn y, Laura Gritti y, Daniel Crooks and Yvonne Dombrowski * Wellcome-Wolfson Institute for Experimental Medicine, Queen’s University Belfast, Belfast BT9 7BL, UK; [email protected] (S.K.); [email protected] (L.G.); [email protected] (D.C.) * Correspondence: [email protected]; Tel.: +0044-28-9097-6127 These authors contributed equally. y Received: 15 October 2019; Accepted: 7 November 2019; Published: 12 November 2019 Abstract: Oligodendrocytes are the myelinating cells of the central nervous system (CNS) that are generated from oligodendrocyte progenitor cells (OPC). OPC are distributed throughout the CNS and represent a pool of migratory and proliferative adult progenitor cells that can differentiate into oligodendrocytes. The central function of oligodendrocytes is to generate myelin, which is an extended membrane from the cell that wraps tightly around axons. Due to this energy consuming process and the associated high metabolic turnover oligodendrocytes are vulnerable to cytotoxic and excitotoxic factors. Oligodendrocyte pathology is therefore evident in a range of disorders including multiple sclerosis, schizophrenia and Alzheimer’s disease. Deceased oligodendrocytes can be replenished from the adult OPC pool and lost myelin can be regenerated during remyelination, which can prevent axonal degeneration and can restore function. Cell population studies have recently identified novel immunomodulatory functions of oligodendrocytes, the implications of which, e.g., for diseases with primary oligodendrocyte pathology, are not yet clear. Here, we review the journey of oligodendrocytes from the embryonic stage to their role in homeostasis and their fate in disease. We will also discuss the most common models used to study oligodendrocytes and describe newly discovered functions of oligodendrocytes. -

Myelin Impairs CNS Remyelination by Inhibiting Oligodendrocyte Precursor Cell Differentiation
328 • The Journal of Neuroscience, January 4, 2006 • 26(1):328–332 Brief Communication Myelin Impairs CNS Remyelination by Inhibiting Oligodendrocyte Precursor Cell Differentiation Mark R. Kotter, Wen-Wu Li, Chao Zhao, and Robin J. M. Franklin Cambridge Centre for Brain Repair and Department of Veterinary Medicine, University of Cambridge, Cambridge CB3 0ES, United Kingdom Demyelination in the adult CNS can be followed by extensive repair. However, in multiple sclerosis, the differentiation of oligodendrocyte lineage cells present in demyelinated lesions is often inhibited by unknown factors. In this study, we test whether myelin debris, a feature of demyelinated lesions and an in vitro inhibitor of oligodendrocyte precursor differentiation, affects remyelination efficiency. Focal demyelinating lesions were created in the adult rat brainstem, and the naturally generated myelin debris was augmented by the addition of purified myelin. After quantification of myelin basic protein mRNA expression from lesion material obtained by laser capture micro- dissection and supported by histological data, we found a significant impairment of remyelination, attributable to an arrest of the differentiation and not the recruitment of oligodendrocyte precursor cells. These data identify myelin as an inhibitor of remyelination as well as its well documented inhibition of axon regeneration. Key words: regeneration; remyelination; oligodendrocyte precursor cell; myelin; differentiation; macrophage Introduction of myelin debris to efficient remyelination and experimental The adult CNS contains a precursor cell population that responds studies in which delayed phagocytosis of myelin debris is associ- to demyelination by undergoing rapid proliferation, migration, ated with a simultaneous delay of OPC differentiation (Kotter et and differentiation into new oligodendrocytes (Horner et al., al., 2005). -
Extensive Remyelination of the CNS Leads to Functional Recovery
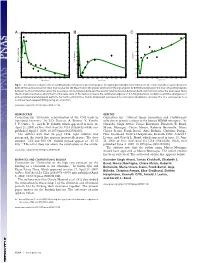
−3 x 10 11 A BC0.35 1 10 0.3 0.9 9 0.25 0.8 0.2 8 0.7 0.15 7 0.6 0.1 0.5 6 Correlation 0.4 0.05 Max. Frequency Max. Power Spectrum 5 0.3 0 4 0.2 −0.05 3 0.1 −0.1 2 0 −0.15 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 2 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 2 0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 2 Noise Level Noise Level Noise Level Fig. 5. Stochastic resonance effects. (A) Maximum of the power spectrum peak of the signal given by differences between the level of synchronization between both communities versus the noise level (variance). (B) Maximum in the power spectrum of the signal given by differences between the level of synchronization between both communities versus the noise level. (C) Correlation between the level of synchronization between both communities versus the noise level. Note the stochastic resonance effect that for the same level of fluctuations reveals the optimal emergence of 0.1-Hz global slow oscillations and the emergence of anticorrelated spatiotemporal patterns for both communities. Points (diamonds) correspond to numerical simulations, whereas the line corresponds to a nonlinear least-squared fitting using an ␣-function. www.pnas.org/cgi/doi/10.1073/pnas.0906701106 NEUROSCIENCE GENETICS Correction for ‘‘Extensive remyelination of the CNS leads to Correction for ‘‘Altered tumor formation and evolutionary functional recovery,’’ by I. -

Lnvited Revie W Growth Factors and Remyelination in The
Histol Histopathol (1997) 12: 459-466 Histology and Histopathology lnvited Revie w Growth factors and remyelination in the CNS R.H. Woodruff and R.J.M. Franklin Departrnent of Clinical Veterinary Medicine and MRC Centre for Brain Repair, University of Carnbridge, Cambridge, UK Summary. It is now well established that there is an multiple sclerosis (MS) (Prineas and Connell, 1979; inherent capacity within the central nervous system Prineas et al., 1989, 1993; Raine and Wu, 1993). (CNS) to remyelinate areas of white matter that have However, remyelination is not universaily consistent or undergone demyelination. However this repair process is sustained, and incomplete remyelination has been not universally consistent or sustained, and persistent reported in gliotoxic lesions of old animals (Gilson and demyelination occurs in a number of situations, most Blakemore, 1993), in certain forms of experimental notably in the chronic multiple sclerosis (MS) plaque. allergic encephalitis (EAE) (Raine et al., 1974), and is a Thus there is a need to investigate ways in which myelin hall-mark feature of the chronic MS plaque. Thus, there deficits within the CNS rnay be restored. One approach is considerable interest in developing ways in which to this problem is to investigate ways in which the persistently demyelinated axons rnay be reinvested with inherent remyelinating capacity of the CNS rnay be myelin sheaths in order to restore secure conduction of stimulated to remyelinate areas of long-term de- impulses. Efforts to address this problem have focussed myelination. The expression of growth factors, which mainly on the use of glial cell transplantation techniques are known to be involved in developmental myelino- to deliver exogenous glial cells into the CNS of hypo- genesis, in areas of demyelination strongly suggests that myelinating myelin mutants (Lachapelle et al., 1984; they are involved in spontaneous remyelination. -

FGF/FGFR Pathways in Multiple Sclerosis and in Its Disease Models
cells Review FGF/FGFR Pathways in Multiple Sclerosis and in Its Disease Models Ranjithkumar Rajendran 1, Gregor Böttiger 1 , Christine Stadelmann 2, Srikanth Karnati 3 and Martin Berghoff 1,* 1 Experimental Neurology, Department of Neurology, University of Giessen, Klinikstrasse 33, 35385 Giessen, Germany; [email protected] (R.R.); [email protected] (G.B.) 2 Institute of Neuropathology, University Medical Center Göttingen, Robert-Koch-Strasse 40, 37075 Göttingen, Germany; [email protected] 3 Institute of Anatomy and Cell Biology, University of Würzburg, Koellikerstrasse 6, 97080 Würzburg, Germany; [email protected] * Correspondence: [email protected]; Tel.: +49-641-98544306; Fax: +49-641-98545329 Abstract: Multiple sclerosis (MS) is a chronic inflammatory and neurodegenerative disease of the central nervous system (CNS) affecting more than two million people worldwide. In MS, oligodendro- cytes and myelin sheaths are destroyed by autoimmune-mediated inflammation, while remyelination is impaired. Recent investigations of post-mortem tissue suggest that Fibroblast growth factor (FGF) signaling may regulate inflammation and myelination in MS. FGF2 expression seems to correlate positively with macrophages/microglia and negatively with myelination; FGF1 was suggested to promote remyelination. In myelin oligodendrocyte glycoprotein (MOG)35–55-induced experimental autoimmune encephalomyelitis (EAE), systemic deletion of FGF2 suggested that FGF2 may promote remyelination. Specific deletion of FGF receptors (FGFRs) in oligodendrocytes in this EAE model resulted in a decrease of lymphocyte and macrophage/microglia infiltration as well as myelin and Citation: Rajendran, R.; Böttiger, G.; axon degeneration. These effects were mediated by ERK/Akt phosphorylation, a brain-derived Stadelmann, C.; Karnati, S.; Berghoff, neurotrophic factor, and downregulation of inhibitors of remyelination. -

The Current Challenges for Drug Discovery in CNS Remyelination
International Journal of Molecular Sciences Review The Current Challenges for Drug Discovery in CNS Remyelination Sonia Balestri, Alice Del Giovane, Carola Sposato, Marta Ferrarelli and Antonella Ragnini-Wilson * Department of Biology, University of Rome “Tor Vergata”, Viale della Ricerca Scientifica, 00133 Rome, Italy; [email protected] (S.B.); [email protected] (A.D.G.); [email protected] (C.S.); [email protected] (M.F.) * Correspondence: [email protected] Abstract: The myelin sheath wraps around axons, allowing saltatory currents to be transmitted along neurons. Several genetic, viral, or environmental factors can damage the central nervous system (CNS) myelin sheath during life. Unless the myelin sheath is repaired, these insults will lead to neurodegeneration. Remyelination occurs spontaneously upon myelin injury in healthy individuals but can fail in several demyelination pathologies or as a consequence of aging. Thus, pharmacological intervention that promotes CNS remyelination could have a major impact on patient’s lives by delaying or even preventing neurodegeneration. Drugs promoting CNS remyelination in animal models have been identified recently, mostly as a result of repurposing phenotypical screening campaigns that used novel oligodendrocyte cellular models. Although none of these have as yet arrived in the clinic, promising candidates are on the way. Many questions remain. Among the most relevant is the question if there is a time window when remyelination drugs should be administrated Citation: Balestri, S.; Del Giovane, and why adult remyelination fails in many neurodegenerative pathologies. Moreover, a significant A.; Sposato, C.; Ferrarelli, M.; challenge in the field is how to reconstitute the oligodendrocyte/axon interaction environment Ragnini-Wilson, A. -

Unexpected Central Role of the Androgen Receptor in the Spontaneous Regeneration of Myelin
Unexpected central role of the androgen receptor in the spontaneous regeneration of myelin Bartosz Bieleckia,b, Claudia Matternc, Abdel M. Ghoumaria, Sumaira Javaida,d, Kaja Smietankaa,b, Charly Abi Ghanema, Sakina Mhaouty-Kodjae, M. Said Ghandourf,g, Etienne-Emile Baulieua,1, Robin J. M. Franklinh,i, Michael Schumachera,1,2, and Elisabeth Traifforta,2 aU1195 INSERM, University Paris-Sud, University Paris-Saclay, Kremlin-Bicêtre 94276, France; bDepartment of Neurology and Stroke, Medical University of Lodz, Lodz 90-549, Poland; cMattern Foundation, Vaduz 9490, Liechtenstein; dHussain Ebrahim Jamal Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi 75270, Pakistan; eU1130 INSERM, UMR 8246 CNRS, University Pierre and Marie Curie, Paris 75005, France; fUMR 7357 CNRS, University of Strasbourg, Strasbourg 67000, France; gDepartment of Anatomy and Neurobiology, Virginia Commonwealth University, Richmond 23284, VA; hWellcome Trust-Medical Research Council Cambridge Stem Cell Institute, University of Cambridge, Cambridge CB2 0AH, United Kingdom; and iDepartment of Clinical Neurosciences, University of Cambridge, Cambridge CB2 0AH, United Kingdom Contributed by Etienne-Emile Baulieu, September 9, 2016 (sent for review June 23, 2016; reviewed by Pamela E. Knapp and Bruce S. McEwen) Lost myelin can be replaced after injury or during demyelinating remyelination was prevented by X-irradiation (4, 6, 7). Unequivocal diseases in a regenerative process called remyelination. In the central confirmation that most Schwann cells contributing to CNS remyeli- nervous system (CNS), the myelin sheaths, which protect axons and nation are indeed derived from OPs has been provided by genetic allow the fast propagation of electrical impulses, are produced by fate-mapping in transgenic mice (5). -

Interaction Between Neurons and the Oligodendroglial Lineage in Multiple Sclerosis and Its Preclinical Models
life Review Interaction between Neurons and the Oligodendroglial Lineage in Multiple Sclerosis and Its Preclinical Models Vasiliki Pantazou 1,2 , Thomas Roux 1,3, Vanessa Oliveira Moreira 1, Catherine Lubetzki 1,3,† and Anne Desmazières 1,*,† 1 Paris Brain Institute (ICM), Sorbonne Université, CNRS, Inserm, GH Pitié-Salpêtrière, 47 boulevard de l’Hôpital, 75013 Paris, France; [email protected] (V.P.); [email protected] (T.R.); [email protected] (V.O.M.); [email protected] (C.L.) 2 Service de Neurologie, Centre Hospitalier Universitaire Vaudois, 46 Rue du Bugnon, 1011 Lausanne, Switzerland 3 Assistance Publique-Hôpitaux de Paris, Neurology Department, Pitié Salpêtrière University Hospital, 75013 Paris, France * Correspondence: [email protected] † These authors contributed equally to this work. Abstract: Multiple sclerosis (MS) is a complex central nervous system inflammatory disease leading to demyelination and associated functional deficits. Though endogenous remyelination exists, it is only partial and, with time, patients can enter a progressive phase of the disease, with neurodegeneration as a hallmark. Though major therapeutic advances have been made, with immunotherapies reducing relapse rate during the inflammatory phase of MS, there is presently no therapy available which significantly impacts disease progression. Remyelination has been shown to favor neuroprotection, and it is thus of major importance to better understand remyelination mechanisms in order to promote them and hence preserve neurons. A crucial point is how this process is regulated through Citation: Pantazou, V.; Roux, T.; the neuronal crosstalk with the oligodendroglial lineage. In this review, we present the current Oliveira Moreira, V.; Lubetzki, C.; knowledge on neuron interaction with the oligodendroglial lineage, in physiological context as well Desmazières, A. -

R-Ras Gtpases Signaling Role in Myelin Neurodegenerative Diseases
International Journal of Molecular Sciences Review R-Ras GTPases Signaling Role in Myelin Neurodegenerative Diseases Berta Alcover-Sanchez , Gonzalo Garcia-Martin , Francisco Wandosell and Beatriz Cubelos * Departamento de Biología Molecular and Centro Biología Molecular “Severo Ochoa”, Universidad Autónoma de Madrid, 28049 Madrid, Spain; [email protected] (B.A.-S.); [email protected] (G.G.-M.); [email protected] (F.W.) * Correspondence: [email protected]; Tel.: +34-91-1964561 Received: 23 July 2020; Accepted: 14 August 2020; Published: 17 August 2020 Abstract: Myelination is required for fast and efficient synaptic transmission in vertebrates. In the central nervous system, oligodendrocytes are responsible for creating myelin sheaths that isolate and protect axons, even throughout adulthood. However, when myelin is lost, the failure of remyelination mechanisms can cause neurodegenerative myelin-associated pathologies. From oligodendrocyte progenitor cells to mature myelinating oligodendrocytes, myelination is a highly complex process that involves many elements of cellular signaling, yet many of the mechanisms that coordinate it, remain unknown. In this review, we will focus on the three major pathways involved in myelination (PI3K/Akt/mTOR, ERK1/2-MAPK, and Wnt/β-catenin) and recent advances describing the crosstalk elements which help to regulate them. In addition, we will review the tight relation between Ras GTPases and myelination processes and discuss its potential as novel elements of crosstalk between the pathways. A better understanding of the crosstalk elements orchestrating myelination mechanisms is essential to identify new potential targets to mitigate neurodegeneration. Keywords: myelin; oligodendrocyte; neurodegeneration; PI3K/Akt/mTOR; ERK1/2-MAPK; Wnt/β-catenin; R-Ras 1. -

Age and Axon-Specific Forms of Cortical Remyelination by Divergent Populations of NG2-Glia
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.09.414755; this version posted December 10, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Age and axon-specific forms of cortical remyelination by divergent populations of NG2-glia Timothy W. Chapman1, Genaro E. Olveda1, Elizabeth Pereira1, Robert A. Hill1,# 1 Department of BiologiCal ScienCes, Dartmouth College, Hanover, NH 03755, USA # corresponding author: [email protected] ABSTRACT Myelin is CritiCal for neural circuit function and its destruction is widespread in neurodegenerative disease and aging. In these Conditions, homeostatic repair meChanisms initiate oligodendrocyte replaCement by resident progenitor cells called NG2-glia. To investigate the Cellular dynamics of this repair we developed a novel demyelination model by combining intravital myelin imaging with a targeted single-cell ablation teChnique Called 2Phatal. OligodendroCyte 2Phatal activated a stereotyped degeneration cascade whiCh triggered remyelination by loCal NG2-glia. Remyelination efficiency was dependent on initial myelin patterning and dynamic imaging revealed rapid repair meChanisms resulting in near-seamless transitions between myelin loss and repair. A subset of morphologiCally Complex NG2-glia executed this remyelination, pointing towards unrecognized funCtional diversity within this population. Age-related demyelination mirrored the degenerative cascade observed with 2Phatal, while remyelination in aging was defective due to failed oligodendrogenesis. Thus, oligodendrocyte 2Phatal revealed Cellular diversity within the oligodendroCyte lineage and uncovered novel forms of rapid remyelination. INTRODUCTION Myelination inCreases neural circuit processing speed and efficiency1,2. -

Human Remyelination Promoting Antibody Stimulates Astrocytes Proliferation Through Modulation of the Sphingolipid Rheostat in Primary Rat Mixed Glial Cultures
Neurochemical Research (2019) 44:1460–1474 https://doi.org/10.1007/s11064-018-2701-x ORIGINAL PAPER Human Remyelination Promoting Antibody Stimulates Astrocytes Proliferation Through Modulation of the Sphingolipid Rheostat in Primary Rat Mixed Glial Cultures Sara Grassi1 · Paola Giussani1 · Simona Prioni1 · Donald Button2 · Jing Cao2 · Irina Hakimi2 · Patrick Sarmiere2 · Maya Srinivas2 · Livia Cabitta1 · Sandro Sonnino1 · Alessandro Prinetti1 Received: 4 August 2018 / Revised: 22 November 2018 / Accepted: 12 December 2018 / Published online: 19 December 2018 © Springer Science+Business Media, LLC, part of Springer Nature 2018 Abstract Remyelination promoting human IgMs effectively increase the number of myelinated axons in animal models of multiple sclerosis. Hence, they ultimately stimulate myelin production by oligodendrocytes (OLs); however, their exact mechanism of action remains to be elucidated, and in particular, it remains unclear whether they are directly targeting OLs, or their action is mediated by effects on other cell types. We assessed the effect of remyelination promoting antibody rHIgM22 on the proliferative response and on the ceramide/sphingosine 1-phosphate rheostat in mixed glial cell cultures (MGCs). rHIgM22 treatment caused a time-dependent increase in PDGFαR protein in MGCs. Forty-eight hours of treatment with rHIgM22 induced a dose-dependent proliferative response (evaluated as total cell number and as EdU(+) cell number) in MGCs. When the proliferation response of MGCs to rHIgM22 was analyzed as a function of the cell types, the most significant proliferative response was associated with GLAST(+) cells, i.e., astrocytes. In many cell types, the balance between different sphingolipid mediators (the “sphingolipid rheostat”), in particular ceramide and sphingosine 1-phosphate, is critical in determining the cell fate. -

Promoting Oligodendrogenesis and Myelin Repair Using the Multiple Sclerosis Medication Glatiramer Acetate
Promoting oligodendrogenesis and myelin repair using the multiple sclerosis medication glatiramer acetate Viktor Skihara, Claudia Silvaa, Andrew Chojnackia, Axinia Do¨ringa, William B. Stallcupb, Samuel Weissa, and V. Wee Yonga,1 aHotchkiss Brain Institute, University of Calgary, Calgary, Alberta T2N 4N1, Canada and bThe Burnham Institute for Medical Research, La Jolla, CA 92037 Communicated by Michael Sela, Weizmann Institute of Science, Rehovot, Israel, August 30, 2009 (received for review May 31, 2009) The formation of oligodendrocytes (oligodendrogenesis) and my- are immunoreactive for neurotrophic factors such as BDNF (7). elin is regulated by several neurotrophic factors. Strategies to However, any approach to use leukocytes to deliver neurotro- increase the level of these trophic molecules may facilitate repair phic factors for repair must balance their potential detriments in in demyelinating conditions, such as multiple sclerosis (MS). Be- exacerbating the pathology of MS. cause leukocytes are a source of neurotrophic factors, and as Glatiramer acetate (GA), a medication used in relapsing- glatiramer acetate (GA) generates T helper 2 (Th2) lymphocytes remitting MS (8), generates GA-reactive T lymphocytes that are that are not known to be harmful, we tested the hypothesis that of the T helper 2 (Th2) anti-inflammatory bias (9, 10). GA- GA regulates oligodendrogenesis and myelin formation. First, we reactive Th2 cells accumulate in the CNS, where they have been generated GA-reactive Th2 cells and determined that they pro- shown to produce not only anti-inflammatory cytokines but also duced transcripts for neurotrophic factors, including insulin-like neurotrophic factors (11). Importantly, Th2 cells have not been growth factor-1 (IGF-1).