Raman Spectroscopy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Comparison of Ultraviolet and Visible Raman Spectra of Supported Metal Oxide Catalysts
8600 J. Phys. Chem. B 2001, 105, 8600-8606 A Comparison of Ultraviolet and Visible Raman Spectra of Supported Metal Oxide Catalysts Yek Tann Chua,† Peter C. Stair,*,† and Israel E. Wachs*,‡ Department of Chemistry, Center for Catalysis and Surface Science and Institute of EnVironmental Catalysis, Northwestern UniVersity, EVanston, Illinois 60208, and Zettlemoyer Center for Surface Studies and Department of Chemical Engineering, Lehigh UniVersity, Bethlehem, PennsylVania 18015 ReceiVed: April 11, 2001 The recent emergence of ultraviolet-wavelength-excited Raman spectroscopy as a tool for catalyst characterization has motivated the question of how UV Raman spectra compare to visible-wavelength-excited Raman spectra on the same catalyst system. Measurements of Raman spectra from five supported metal oxide systems (Al2O3-supported Cr2O3,V2O5, and MoO3 as well as TiO2-supported MoO3 and Re2O7), using visible (514.5 nm) and ultraviolet (244 nm) wavelength excitation have been compared to determine the similarities and differences in Raman spectra produced at the two wavelengths. The samples were in the form of self-supporting disks. Spectra from the oxides, both hydrated as a result of contact with ambient air and dehydrated as a result of calcination or laser-induced heating, were recorded. A combination of sample spinning and translation to produce a spiral pattern of laser beam exposure to the catalyst disk was found to be most effective in minimizing dehydration caused by laser-induced heating. Strong absorption by the samples in the ultraviolet significantly reduced the number of scatterers contributing to the Raman spectrum while producing only modest increases in the Raman scattering cross section due to resonance enhancement. -
CHAPTER 13 Molecular Spectroscopy 2: Electronic Transitions
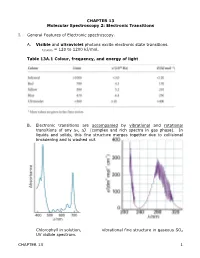
CHAPTER 13 Molecular Spectroscopy 2: Electronic Transitions I. General Features of Electronic spectroscopy. A. Visible and ultraviolet photons excite electronic state transitions. εphoton = 120 to 1200 kJ/mol. Table 13A.1 Colour, frequency, and energy of light B. Electronic transitions are accompanied by vibrational and rotational transitions of any Δν, ΔJ (complex and rich spectra in gas phase). In liquids and solids, this fine structure merges together due to collisional broadening and is washed out. Chlorophyll in solution, vibrational fine structure in gaseous SO2 UV visible spectrum. CHAPTER 13 1 C. Molecules need not have permanent dipole to undergo electronic transitions. All that is required is a redistribution of electronic and nuclear charge between the initial and final electronic state. Transition intensity ~ |µ|2 where µ is the magnitude of the transition dipole moment given by: ˆ d µ = ∫ ψfinalµψ initial τ where µˆ = −e ∑ri + e ∑Ziri € electrons nuclei r vector position of the particle i = D. Franck-Condon principle: Because nuclei are so much more massive and € sluggish than electrons, electronic transitions can happen much faster than the nuclei can respond. Electronic transitions occur vertically on energy diagram at right. (Hence the name vertical transition) Transition probability depends on vibrational wave function overlap (Franck-Condon factor). In this example: ν = 0 ν’ = 0 have small overlap ν = 0 ν’ = 2 have greatest overlap Now because the total molecular wavefunction can be approximately factored into 2 terms, electronic wavefunction and the nuclear position wavefunction (using Born-Oppenheimer approx.) the transition dipole can also be factored into 2 terms also: S µ = µelectronic i,f S d Franck Condon factor i,j i,f = ∫ ψf,vibr ψi,vibr τ = − CHAPTER 13 2 See derivation “Justification 14.2” II. -

Atomic and Molecular Laser-Induced Breakdown Spectroscopy of Selected Pharmaceuticals
Article Atomic and Molecular Laser-Induced Breakdown Spectroscopy of Selected Pharmaceuticals Pravin Kumar Tiwari 1,2, Nilesh Kumar Rai 3, Rohit Kumar 3, Christian G. Parigger 4 and Awadhesh Kumar Rai 2,* 1 Institute for Plasma Research, Gandhinagar, Gujarat-382428, India 2 Laser Spectroscopy Research Laboratory, Department of Physics, University of Allahabad, Prayagraj-211002, India 3 CMP Degree College, Department of Physics, University of Allahabad, Pragyagraj-211002, India 4 Physics and Astronomy Department, University of Tennessee, University of Tennessee Space Institute, Center for Laser Applications, 411 B.H. Goethert Parkway, Tullahoma, TN 37388-9700, USA * Correspondence: [email protected]; Tel.: +91-532-2460993 Received: 10 June 2019; Accepted: 10 July 2019; Published: 19 July 2019 Abstract: Laser-induced breakdown spectroscopy (LIBS) of pharmaceutical drugs that contain paracetamol was investigated in air and argon atmospheres. The characteristic neutral and ionic spectral lines of various elements and molecular signatures of CN violet and C2 Swan band systems were observed. The relative hardness of all drug samples was measured as well. Principal component analysis, a multivariate method, was applied in the data analysis for demarcation purposes of the drug samples. The CN violet and C2 Swan spectral radiances were investigated for evaluation of a possible correlation of the chemical and molecular structures of the pharmaceuticals. Complementary Raman and Fourier-transform-infrared spectroscopies were used to record the molecular spectra of the drug samples. The application of the above techniques for drug screening are important for the identification and mitigation of drugs that contain additives that may cause adverse side-effects. Keywords: paracetamol; laser-induced breakdown spectroscopy; cyanide; carbon swan bands; principal component analysis; Raman spectroscopy; Fourier-transform-infrared spectroscopy 1. -

Laser Raman Spectroscopy As a Technique for Identification Of
ARTICLE IN PRESS CHEMGE-15589; No of Pages 13 Chemical Geology xxx (2008) xxx–xxx Contents lists available at ScienceDirect Chemical Geology journal homepage: www.elsevier.com/locate/chemgeo Laser Raman spectroscopy as a technique for identification of seafloor hydrothermal and cold seep minerals Sheri N. White ⁎ Department of Applied Ocean Physics and Engineering, Woods Hole Oceanographic Institution, Woods Hole, MA 02536, USA article info abstract Article history: In situ sensors capable of real-time measurements and analyses in the deep ocean are necessary to fulfill the Received 8 August 2008 potential created by the development of autonomous, deep-sea platforms such as autonomous and remotely Received in revised form 8 November 2008 operated vehicles, and cabled observatories. Laser Raman spectroscopy (a type of vibrational spectroscopy) is an Accepted 10 November 2008 optical technique that is capable of in situ molecular identification of minerals in the deep ocean. The goals of this Available online xxxx work are to determine the characteristic spectral bands and relative Raman scattering strength of hydrothermally- Editor: R.L. Rudnick and cold seep-relevant minerals, and to determine how the quality of the spectra are affected by changes in excitation wavelength and sampling optics. The information learned from this work will lead to the development Keywords: of new, smaller sea-going Raman instruments that are optimized to analyze minerals in the deep ocean. Raman spectroscopy Many minerals of interest at seafloor hydrothermal and cold seep sites are Raman active, such as elemental sulfur, Mineralogy carbonates, sulfates and sulfides. Elemental S8 sulfur is a strong Raman scatterer with dominant bands at ∼219 and Hydrothermal vents 472 Δcm−1. -

Accessing Excited State Molecular Vibrations by Femtosecond Stimulated Raman Spectroscopy
Accessing Excited State Molecular Vibrations by Femtosecond Stimulated Raman Spectroscopy Giovanni Batignani,y Carino Ferrante,y,z and Tullio Scopigno∗,y,z yDipartimento di Fisica, Universitá di Roma “La Sapienza", Roma, I-00185, Italy z Istituto Italiano di Tecnologia, Center for Life Nano Science @Sapienza, Roma, I-00161, Italy E-mail: [email protected] arXiv:2010.05029v1 [physics.optics] 10 Oct 2020 1 Abstract Excited-state vibrations are crucial for determining photophysical and photochem- ical properties of molecular compounds. Stimulated Raman scattering can coherently stimulate and probe molecular vibrations with optical pulses, but it is generally re- stricted to ground state properties. Working in resonance conditions, indeed, enables cross-section enhancement and selective excitation to a targeted electronic level, but is hampered by an increased signal complexity due to the presence of overlapping spectral contributions. Here, we show how detailed information on ground and excited state vi- brations can be disentangled, by exploiting the relative time delay between Raman and probe pulses to control the excited state population, combined with a diagrammatic formalism to dissect the pathways concurring to the signal generation. The proposed method is then exploited to elucidate the vibrational properties of ground and excited electronic states in the paradigmatic case of Cresyl Violet. We anticipate that the presented approach holds the potential for selective mapping the reaction coordinates pertaining to transient electronic stages implied in photo-active compounds. Graphical TOC Entry 2 Raman spectroscopy is a powerful tool to access the vibrational fingerprints of molecules or solid state compounds and it can be used to extract structural and dynamical information of the samples under investigation. -

Combining Chemical Information from Grass Pollen in Multimodal Characterization
ORIGINAL RESEARCH published: 31 January 2020 doi: 10.3389/fpls.2019.01788 Combining Chemical Information From Grass Pollen in Multimodal Characterization Sabrina Diehn 1,2, Boris Zimmermann 3, Valeria Tafintseva 3, Stephan Seifert 1,2, Murat Bag˘ cıog˘ lu 3, Mikael Ohlson 4, Steffen Weidner 2, Siri Fjellheim 5, Achim Kohler 3,6 and Janina Kneipp 1,2* 1 Department of Chemistry, Humboldt-Universität zu Berlin, Berlin, Germany, 2 BAM Federal Institute for Materials Research and Testing, Berlin, Germany, 3 Faculty of Science and Technology, Norwegian University of Life Sciences, Ås, Norway, 4 Faculty of Environmental Sciences and Natural Resource Management, Norwegian University of Life Sciences, Ås, Norway, 5 Faculty of Biosciences, Norwegian University of Life Sciences, Ås, Norway, 6 Nofima AS, Ås, Norway Edited by: Lisbeth Garbrecht Thygesen, fi University of Copenhagen, The analysis of pollen chemical composition is important to many elds, including Denmark agriculture, plant physiology, ecology, allergology, and climate studies. Here, the Reviewed by: potential of a combination of different spectroscopic and spectrometric methods Wesley Toby Fraser, regarding the characterization of small biochemical differences between pollen samples Oxford Brookes University, United Kingdom was evaluated using multivariate statistical approaches. Pollen samples, collected from Åsmund Rinnan, three populations of the grass Poa alpina, were analyzed using Fourier-transform infrared University of Copenhagen, Denmark (FTIR) spectroscopy, Raman spectroscopy, surface enhanced Raman scattering (SERS), Anna De Juan, and matrix assisted laser desorption/ionization mass spectrometry (MALDI-TOF MS). The University of Barcelona, Spain variation in the sample set can be described in a hierarchical framework comprising three *Correspondence: populations of the same grass species and four different growth conditions of the parent Janina Kneipp [email protected] plants for each of the populations. -

Raman Scattering and Fluorescence
Fluorescence 01 Raman Scattering and Fluorescence Introduction The existence of such virtual states also explains why the non-resonance Raman effect Raman scattering and Fluorescence emission does not depend on the wavelength of the are two competing phenomena, which have excitation, since no real states are involved in similar origins. Generally, a laser photon this interaction mechanism. In fact, the Raman bounces off a molecule and looses a certain spectrum generally does not depend on the amount of energy that allows the molecule to laser excitation. vibrate (Stokes process). The scattered photon is therefore less energetic and the associated However, when the energy of the excitation light exhibits a frequency shift. The various photon gets close to the transition energy frequency shifts associated with different between two electronic states, one then deals molecular vibrations give rise to a spectrum, with resonance Raman or resonance that is characteristic of a specific compound. fluorescence (fig.1, case (d)). The basic difference between these two processes is In contrast, fluorescence or luminescence related to the time scales involved, as well as emission follows an absorption process. For a with the nature of the so-called intermediate better understanding, one can refer to the states. In contrast with resonant fluorescent, diagram below. relaxed fluorescence results from the emission of a photon from the lowest vibrational level of an excited electronic state, following a direct absorption of the photon and relaxation of the molecule from its vibrationally excited level of the electronic state back to the lowest vibrational level of the electronic state. A fluorescence process typically requires more than 10-9 s. -

Lecture B4 Vibrational Spectroscopy, Part 2 Quantum Mechanical SHO
Lecture B4 Vibrational Spectroscopy, Part 2 Quantum Mechanical SHO. WHEN we solve the Schrödinger equation, we always obtain two things: 1. a set of eigenstates, |ψn>. 2. a set of eigenstate energies, En QM predicts the existence of discrete, evenly spaced, vibrational energy levels for the SHO. n = 0,1,2,3... For the ground state (n=0), E = ½hν. Notes: This is called the zero point energy. Optical selection rule -- SHO can absorb or emit light with a ∆n = ±1 The IR absorption spectrum for a diatomic molecule, such as HCl: diatomic Optical selection rule 1 -- SHO can absorb or emit light with a ∆n = ±1 Optical selection rule 2 -- a change in molecular dipole moment (∆μ/∆x) must occur with the vibrational motion. (Note: μ here means dipole moment). If a more realistic Morse potential is used in the Schrödinger Equation, these energy levels get scrunched together... ΔE=hν ΔE<hν The vibrational spectroscopy of polyatomic molecules gets more interesting... For diatomic or linear molecules: 3N-5 modes For nonlinear molecules: 3N-6 modes N = number of atoms in molecule The vibrational spectroscopy of polyatomic molecules gets more interesting... Optical selection rule 1 -- SHO can absorb or emit light with a ∆n = ±1 Optical selection rule 2 -- a change in molecular dipole moment (∆μ/∆x) must occur with the vibrational motion of a mode. Consider H2O (a nonlinear molecule): 3N-6 = 3(3)-6 = 3 Optical selection rule 2 -- a change in molecular dipole moment (∆μ/∆x) must occur with the vibrational motion of a mode. Consider H2O (a nonlinear molecule): 3N-6 = 3(3)-6 = 3 All bands are observed in the IR spectrum. -

Technical Application Note
Compact Rugged Spectrometers - A Universe of Spectroscopy Systems Technical Application Note Vibrational Spectroscopy: Infrared vs. Raman StellarNet, Inc. Tampa, Fl USA Tony Rizzuto, PhD April 12, 2017 Vibrational spectroscopies are integral in analyzing some of the most fundamentally important processes in physical chemistry: molecular vibrations. While there are many different experimental techniques used to analyze those vibrations, most are variations of the “Big Two,” FTIR and Raman scattering spectroscopies. This article aims to analyze the benefits and drawbacks of each of the “Big Two,” with the hope that it helps one choose the correct technique for one’s research interests. 1. What are the “Big Two?” Fourier Transform Infrared Spectroscopy Raman spectroscopy relies on inelastic (FTIR) is a simple absorption measurement scattering phenomenon that probes the where the detector measures the molecular vibration. Where FTIR will use a absorbance of infrared radiation by the broadband IR source, Raman spectroscopy sample. Each sample will absorb different typically uses a narrow-band, amounts of each frequency resulting in a monochromatic light source in order to “chemical fingerprint” that is the FTIR excite the vibrations of the molecule in your spectrum. sample. 2. Selection Rules: What molecular vibrations are being probed? Imagining a molecular bond vibration with the traditional ball and spring model (Figure 1a) and its resultant harmonic oscillator depiction (Figure 1b) allows us to calculate the selection rules for vibrational transitions to be = 1. However, that does not tell the whole story. In order to truly differentiate FTIR from Raman Spectroscopy, we must think about it on a molecular level. Compact Rugged Spectrometers - A Universe of Spectroscopy Systems Technical Application Note A B Figure 1: A. -

Lecture 17 Highlights
Lecture 23 Highlights The phenomenon of Rabi oscillations is very different from our everyday experience of how macroscopic objects (i.e. those made up of many atoms) absorb electromagnetic radiation. When illuminated with light (like French fries at McDonalds) objects tend to steadily absorb the light and heat up to a steady state temperature, showing no signs of oscillation with time. We did a calculation of the transition probability of an atom illuminated with a broad spectrum of light and found that the probability increased linearly with time, leading to a constant transition rate. This difference comes about because the many absorption processes at different frequencies produce a ‘smeared-out’ response that destroys the coherent Rabi oscillations and results in a classical incoherent absorption. Selection Rules All of the transition probabilities are proportional to “dipole matrix elements,” such as x jn above. These are similar to dipole moment calculations for a charge distribution in classical physics, except that they involve the charge distribution in two different states, connected by the dipole operator for the transition. In many cases these integrals are zero because of symmetries of the associated wavefunctions. This gives rise to selection rules for possible transitions under the dipole approximation (atom size much smaller than the electromagnetic wave wavelength). For example, consider the matrix element z between states of the n,l , m ; n ', l ', m ' Hydrogen atom labeled by the quantum numbers n,,l m and n',','l m : z = ψ()()r z ψ r d3 r n,l , m ; n ', l ', m ' ∫∫∫ n,,l m n',l ', m ' To make progress, first consider just the φ integral. -

Article Intends to Provide a for the Necessary Virtual Electronic Brief Overview of the Differences and Transition
ADVANCES IN RAMAN TECHNIQUES Laser requirements and advances for Raman techniques Andreas Isemann Laser Quantum GmbH, 78467 Konstanz, Germany INTRODUCTION 473 nm and 1064 nm, a narrow Raman scattering as a probe of bandwidth output of few tens of GHz vibrational transitions has made or below 1 MHz if needed within the leaps and bounds since its discovery, linewidth of vibrational transitions and various schemes based on this for high resolution, low noise (less phenomenon have been developed than 0.02%) and excellent beam with great success. quality (fundamental transversal Applications range from basic electromagnetic mode TEM00) scientific research, to medical and provides optimised performance industrial instrumentation. Some for the resolution of the Raman schemes utilise linear Raman measurement needed. scattering, whilst others take advantage The wavelength is chosen based of high peak-power fields to probe on the sample under investigation, nonlinear Raman responses. with 532 nm being commonly used This article intends to provide a for the necessary virtual electronic brief overview of the differences and transition. In the following section, benefits, together with the laser source four examples from different areas of requirements and the advancements Raman applications show the diverse in techniques enabled by recent applications of linear Raman and what developments in lasers. advances have been achieved. An example of studying a real-world LINEAR RAMAN application, the successful control of Figure 1 An example of the RR microfluidic device counting of The advent of the laser in providing a food quality using Raman spectroscopy photosynthetic microorganisms. As the cells of the model strain high-intensity coherent light source and multivariate analysis, is described Synechocystis sp. -

Introduction to Differential Equations
Quantum Dynamics – Quick View Concepts of primary interest: The Time-Dependent Schrödinger Equation Probability Density and Mixed States Selection Rules Transition Rates: The Golden Rule Sample Problem Discussions: Tools of the Trade Appendix: Classical E-M Radiation POSSIBLE ADDITIONS: After qualitative section, do the two state system, and then first and second order transitions (follow Fitzpatrick). Chain together the dipole rules to get l = 2,0,-2 and m = -2, -2, … , 2. ??Where do we get magnetic rules? Look at the canonical momentum and the Asquared term. Schrödinger, Erwin (1887-1961) Austrian physicist who invented wave mechanics in 1926. Wave mechanics was a formulation of quantum mechanics independent of Heisenberg's matrix mechanics. Like matrix mechanics, wave mechanics mathematically described the behavior of electrons and atoms. The central equation of wave mechanics is now known as the Schrödinger equation. Solutions to the equation provide probability densities and energy levels of systems. The time-dependent form of the equation describes the dynamics of quantum systems. http://scienceworld.wolfram.com/biography/Schroedinger.html © 1996-2006 Eric W. Weisstein www-history.mcs.st-andrews.ac.uk/Biographies/Schrodinger.html Quantum Dynamics: A Qualitative Introduction Introductory quantum mechanics focuses on time-independent problems leaving Contact: [email protected] dynamics to be discussed in the second term. Energy eigenstates are characterized by probability density distributions that are time-independent (static). There are examples of time-dependent behavior that are by demonstrated by rather simple introductory problems. In the case of a particle in an infinite well with the range [ 0 < x < a], the mixed state below exhibits time-dependence.