The Device Physics of Organic Solar Cells a Dissertation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 5 Semiconductor Laser
Chapter 5 Semiconductor Laser _____________________________________________ 5.0 Introduction Laser is an acronym for light amplification by stimulated emission of radiation. Albert Einstein in 1917 showed that the process of stimulated emission must exist but it was not until 1960 that TH Maiman first achieved laser at optical frequency in solid state ruby. Semiconductor laser is similar to the solid state laser like the ruby laser and helium-neon gas laser. The emitted radiation is highly monochromatic and produces a highly directional beam of light. However, the semiconductor laser differs from other lasers because it is small in 0.1mm long and easily modulated at high frequency simply by modulating the biasing current. Because of its uniqueness, semiconductor laser is one of the most important light sources for optical-fiber communication. It can be used in many other applications like scientific research, communication, holography, medicine, military, optical video recording, optical reading, high speed laser printing etc. The analysis of physics of laser is quite difficult and we summarize with the simplified version here. The application of laser although with a slow start in the 1960s but now very often new applications are found such as those mentioned earlier in the text. 5.1 Emission and Absorption of Radiation As mentioned in earlier Chapter, when an electron in an atom undergoes transition between two energy states or levels, it either absorbs or emits photon. When the electron transits from lower energy level to higher energy level, it absorbs photon. When an electron transits from higher energy level to lower energy level, it releases photon. -

Population Inversion in Atoms, Molecules, and Semiconductors Atoms and Molecules
OPTI 500 DEF, Spring 2012, Lecture 2 Introduction to Sources: Radiative Processes and Population Inversion in Atoms, Molecules, and Semiconductors Atoms and Molecules Energy Levels Every atom or molecule has a collection of discrete energy levels that may be irregularly spaced. Figure 1 shows six levels in a hypothetical collection. Each of the energy levels may be comprised of multiple sublevels of the same energy, in which case we say that the energy level is degenerate. For the time being we will ignore degeneracy. As pictured in Figure 1, the atom or molecule is in level 1. 6 6 5 5 4 4 3 3 2 2 1 1 Level 0 Level 0 Figure 1. Energy levels for an atom or molecule. Frequently we will discuss the interaction of light with just two of the levels. Monochromatic light generated by a laser may interact strongly with only two levels, say levels 1 and 2, if the photon energy equals the 1/19 Radiative Processes and Population Inversion difference in the energy of the two levels (i.e. hν = E2 – E1). In this case we say that the light interacts resonantly with levels 1 and 2, and we often focus our attention on these levels and, at least temporarily, ignore the rest. From here on we will refer to an atom or molecule as a particle. We almost always deal with a collection of particles, each of which may be in a different energy level as pictured in Figure 2. Because the number of particles may be very large, it is common to use a visual shorthand and represent the collection with just two levels as in Figure 3. -

3 Electrons and Holes in Semiconductors
3 Electrons and Holes in Semiconductors 3.1. Introduction Semiconductors : optimum bandgap Excited carriers → thermalisation Chap. 3 : Density of states, electron distribution function, doping, quasi thermal equilibrium electron and hole currents Chap. 4 : Charge carrier generation, recombination, transport equation 3.2. Basic Concepts 3.2.1. Bonds and bands in crystals Free electron approximation to band Ref. CLASSIC “Bonds and Bands in Semiconductors” J. C. Phillips, Academic 1973 Atomic orbitals → molecular orbitals → bands Valence band : HOMO Conduction band : LUMO 1 Semiconductors : 0.5 Eg 3 eV Semi-metals : 0 Eg 0.5 eV Si : sp3 orbital, diamond-like structure 3.2.2. Electrons, holes and conductivity Semiconductors At T 0 K, all electrons are in valence band – no conductivity At elevated temperature, some electrons in conduction band, and some holes in valence band 2 Conduction Band Electrons Valence Band a) a)’ Electron in CB Hole in VB b) b)’ Fig. 3.4. Electron in CB and Hole in VB 3.3. Electron States in Semiconductors 3.3.1. Band structure Electronic states in crystalline Solving Schrödinger equation in periodic potential (infinite) Bloch wavefunction ikr k, r uik r e (3.1) for crystal band i and a wavevector k, which is a good quantum number. The energy E against wavevector k is called by crystal band structure. Typical band diagram plotted along the 3 major directions in the reciprocal space. 0 0 0, X 10 0, M110, L111 3 Point k is called the Brillouin zone boundary. a 2 3 3 For zinc blende structure, 0 0 0, X 0 0, K 0, L a 2a 2a a a a 3.3.2. -

University Microfilms, Inc., Ann Arbor, Michigan APPLICATION of the SHOCKLEY-READ RECOMBINATION
This dissertation has been 64—6944 microfilmed exactly as received PANG, Tet Chong, 1929- APPLICATION OF THE SHOCKLEY-READ RECOMBINATION STATISTICS TO THE STUDY OF THE P*NN+ DIODE. The Ohio State University, Ph.D., 1963 Engineering, electrical University Microfilms, Inc., Ann Arbor, Michigan APPLICATION OF THE SHOCKLEY-READ RECOMBINATION STATISTICS TO THE STUDY OF THE P+NN+ DIODE DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Ftiilosophy in the Graduate School of The Ohio State lhiversity By Tet Chong Pang, B.S., M.Sc. The Ohio State lhiversity 1963 Approved by Adviser Department of Electrical Engineering PLEASE NOTE: Figures are not original copy. Pages tend to "curl". Filmed in the best possible way. UNIVERSITY MICROFILMS, INC. ACKNOWLEDGMENTS This research was started when the author was a student of Professor E. Milton Boone to whom he is ever grateful for his overall guidance and understanding over the years. He is deeply indebted to Professor Marlin 0. Thurston for his encouragement and many elucidating discussions. It is a pleasure to acknowledge the assistance in computer programming given by the staff of the Numerical Computation Laboratory, The Ohio State Lhiversity, in particular, Professor Theodore W. Hildebrandt. CONTENTS Page ACKNOWLEDGMENTS..................................... ii LIST OF ILLUSTRATIONS................................ v Chapter I INTRODUCTION ............................... 1 II RECOMBINATION OF ELECTRONS AND HOLES ........... 3 III SHOCKLEY-READ RECOMBINATION STATISTICS ......... 6 IV DETERMINATION OF CARRIER DENSITIES ............ 12 V CONDUCTIVITY MODULATION AND NEGATIVE RESISTANCE .............................. 17 Conductivity modulation ................... 17 Negative resistance ...................... 17 Lifetime variation with carrier density .... 18 VI ANALYTICAL APPROACH TO THE SOLUTION OF TRANSPORT EQUATION ...................... 24 VII NUMERICAL APPROACH TO THE SOLUTION OF TRANSPORT E Q U A T I O N ..................... -

Quasi-Fermi Levels
6.772/SMA5111 - Compound Semiconductors Supplement 1 - Semiconductor Physics Review - Outline • The Fermi function and the Fermi level The occupancy of semiconductor energy levels • Effective density of states Conduction and valence band density of states 1. General 2. Parabolic bands • Quasi Fermi levels The concept and definition Examples of application 1. Uniform electric field on uniform sample 2. Forward biased p-n junction 3. Graded composition p-type heterostructure 4. Band edge gradients as effective forces for carrier drift Refs: R. F. Pierret, Semiconductor Fundamentals 2nd. Ed., (Vol. 1 of the Modular Series on Solid State Devices, Addison-Wesley, 1988); TK7871.85.P485; ISBN 0-201-12295-2. S. M. Sze, Physics of Semiconductor Devices (see course bibliography) Appendix C in Fonstad (handed out earlier; on course web site) C. G. Fonstad, 2/03 Supplement 1 - Slide 1 Fermi level and quasi-Fermi Levels - review of key points Fermi level: In thermal equilibrium the probability of finding an energy level at E occupied is given by the Fermi function, f(E): - f (E) =1 (1 +e[E E f ]/ kT ) where Ef is the Fermi energy, or level. In thermal equilibrium Ef is constant and not a function of position. The Fermi function has the following useful properties: - - ª [E E f ]/ kT - >> f (E) e for (E E f ) kT - ª - [E E f ]/ kT - << - f (E) 1 e for (E E f ) kT = = f (E f ) 1/2 for E E f These relationships tell us that the population of electrons decreases exponentially with energy at energies much more than kT above the Fermi level, and similarly that the population of holes (empty electron states) decreases exponentially with energy when more than kT below the Fermi level. -
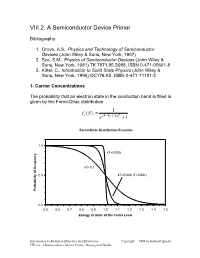
VIII.2. a Semiconductor Device Primer
VIII.2. A Semiconductor Device Primer Bibliography: 1. Grove, A.S., Physics and Technology of Semiconductor Devices (John Wiley & Sons, New York, 1967) 2. Sze, S.M., Physics of Semiconductor Devices (John Wiley & Sons, New York, 1981) TK 7871.85.S988, ISBN 0-471-05661-8 3. Kittel, C., Introduction to Solid State Physics (John Wiley & Sons, New York, 1996) QC176.K5, ISBN 0-471-11181-3 1. Carrier Concentrations The probability that an electron state in the conduction band is filled is given by the Fermi-Dirac distribution = 1 fe (E) − e(E EF )/kBT +1 Fermi-Dirac Distribution Function 1.0 kT=0.005 kT=0.1 0.5 kT=0.026 (T=300K) Probability of Ocupancy 0.0 0.5 0.6 0.7 0.8 0.9 1.0 1.1 1.2 1.3 1.4 1.5 Energy in Units of the Fermi Level Introduction to Radiation Detectors and Electronics Copyright 1998 by Helmuth Spieler VIII.2.a. A Semiconductor Device Primer, Doping and Diodes The density of atoms in a Si or Ge crystal is about 4.1022 atoms/cm3. Since the minimum carrier density of interest in practical devices is of order 1010 to 1011 cm-3, very small ocupancy probabilities are quite important. Fermi-Dirac Distribution Function 1.0E+00 1.0E-02 1.0E-04 1.0E-06 kT=0.035 eV (T=400K) 1.0E-08 kT=0.026 eV (T=300K) 1.0E-10 1.0E-12 Probability of Occupancy 1.0E-14 1.0E-16 1.0E-18 0.8 0.9 1 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 1.9 2 Energy in Units of Fermi Level In silicon the band gap is 1.12 eV. -

Law of the Junction Revisited (Lundstrom)
The Law of the Junction Revisited Mark Lundstrom Network for Computational Nanotechnology and Purdue University Consider a one-sided, short base diode like that shown in Fig. 1. We usually analyze the I-V characteristics by assuming the so-called Law of the Junction, n 2 n(0) n eqVA kBT 1 i eqVA kBT 1 ! = po( " ) = ( " ). (1) NA We find the current by assuming that electrons diffuse across the p-region, so !n(0) D n 2 J qD q n i eqVA kBT 1 n = n = ( " ). (2) WP WP NA (If the diode has a long p-type region, then WP is replaced by Ln , the minority carrier diffusion length.) To justify the Law of the Junction, we assume low level injection and that the quasi- Fermi levels are constant across the depletion region, as sketched in Fig. 1. EC Fn qVA Fp EV xp xp WP+xp Fig.1 An NP homojunction under forward bias The Law of the Junction generally works well, but under high bias, the assumptions of constant quasi-Fermi levels across the space-charge region and low level injection in the p-region may begin to lose validity. Lundstrom 1 2/27/13 The metal-semiconductor (MS) diode sketched in Fig. 2 is a different kind of junction. The Law of the Junction cannot be used in this case, so its I-V characteristics must be derived differently. q V V ( bi ! A ) EC φBn qV Fn A EFM x 0 Fig. 2 A metal-semiconductor (MS) diode under forward bias. Most MS junctions obey the thermionic emission theory. -

A-Guide-To-Blackbody-Physics.Pdf
A guide to blackbody physics Daniel Suchet∗ 2017 - 12 - 08 Short summary Section 1 Radiometrics This part is quite boring, but it is necessary to well define the quantities at stake : their definition are non-intuitive at all and lead to easy mistakes. Moreover, many authors have their own definition / notations of some quantities, making it even more confusing. The following quantities are considered in this section: Radiant flux, Radiant exitance, Irradiance, Radiant intensity, Radiance, Optical étendue, Absorption coefficient, Absorptivity and Absorption rate Section 2 Thermodynamics of a (blackbody) radiation in a cavity The photon density inside a blackbody (or gray-body) is derived, using several methods. No- tably, the van Roosbroeck - Shockley equation, expressing the spontaneous emission rate inside the medium is obtained. Section 3 Thermodynamics of a blackbody radiation in free space A discussion on the relation between the radiation inside the body and the radiation emitted by the body is offered and emitted flux of photon, energy and entropy are calculated. Several tricky ques- tions are addressed: what is the entropy produced by emitting radiation ? What does a blackbody look like ? What is the validity range of classical approximations ? Should étendue or aperture be considered ? Section 4 Thermodynamics of graybodies Kirchhoff law of radiation and the Shockley Queisser limit are presented in details, including a long discussion on chemical potentials in solar cells. The approach of Tom Markvart is also presented to give a thermodynamic interpretation on energy loss terms. Contents 1 Radiometrics 2 1.1 Radiometric quantities . 2 1.2 Optical étendue . 3 1.3 Absorption coefficient, absorptivity, absorption rate . -

Quasi Fermi Level Scan of Band Gap Energy in Photojunction B.A
Vol. 134 (2018) ACTA PHYSICA POLONICA A No. 2 Quasi Fermi Level Scan of Band Gap Energy in Photojunction B.A. Orłowskia;∗, K. Gwóźdźb, M. Galickaa, S. Chusnutdinowa, E. Placzek-Popkob, M.A. Pietrzyka, E. Guziewicza and B.J. Kowalskia aInstitute of Physics, Polish Academy of Sciences, Aleja Lotnikow 32/46, PL-02668 Warsaw, Poland bDepartment of Quantum Technology, Faculty of Fundamental Problems of Technology, Wroclaw University of Science and Technology, Wybrzeze Wyspianskiego 27, 50-370 Wroclaw, Poland (Received April 9, 2018; in final form June 28, 2018) Photovoltage generation model results are compared with the correlated illumination intensity spectra of semiconductors photojunction. The moderate continuous increase of illumination intensity of semiconductor pho- tojunction leads to remarkable increase of relative concentration of minority carriers and related to it quasi Fermi level scan along the energy band gap. The scanning energy region runs up from thermal equilibrium Fermi level for electrons and down for holes. For moderate illumination related changes of quasi Fermi levels energy of mi- nority carriers dominate over the changes of majority carriers and they decide on measured open circuit voltage. Expected spectrum of quasi Fermi level scan on illumination intensity will strongly depend on interaction with electronic “defects” located in photojunction region (e.g. impurities, clusters, barriers, etc.) leading to the major- ity quasi Fermi level pinning. Measured region of quasi Fermi level energy pinning allows to estimate the defect states parameters (binding energy and concentration) in situ during the work of photojunction. The theoretical model of described effect will be presented and supported by experimental data measured for Si p=n junction and CdTe/ZnTe heterojunction. -

Indian Institute of Technology Roorkee
INDIAN INSTITUTE OF TECHNOLOGY ROORKEE NAME OF DEPTT./CENTRE: DEPARTMENT OF PHYSICS 1. Subject Code: PH-701 Course Title: Laboratory 2. Contact Hours: L: 0 T: 0 P: 6 3. Examination Duration (Hrs.): Theory 0 Practical 6 4. Relative Weightage: CWS 1 0 PRS MTE50 ETE0 PRE0 50 5. Credits: 3 6. Semester: Autumn 7. Subject Area: PCC 8. Pre-requisite: Nil 9. Objective: To impart practical knowledge in Solid Sate Electronic Materials 10. Details of Course: S. No. Contact Contents Hours 1. Study of variation of resistivity with temperature of metal and highly resistive materials by Four Probe Technique. 2. Mapping and analysis of the resistivity of large samples (thin films, superconductors) by Four probe Technique. 3. To study the temperature dependence of Hall coefficient of n- and p- type semiconductors. 4. (a) To measure the dielectric constant and Curie temperature of given ferroelectric samples. (b) To measure the coercive field (Ec), remanent polarization (Pr), Curie temperature (Tc) and spontaneous polarization (Ps) of Barium Titanate (BaTiO3). 14 x 6 5. Thermoluminescence in alkali halides crystals. (a) To produce F centers in the crystal exposing to X-ray /UV source. (b) To determine activation energy of the F-centers by initial rise method. 6. Verification of Bragg’s law and determination of wavelength/energy spectrum of X-rays. 7. Study of solar cell characteristics and to determine open circuit voltage ‘Voc’ , short circuit current ‘Isc’, Efficiency ( ), fill factor, spectral characteristics and chopper characteristics. 8. To measure the magnetoresistance of semiconductor and analyze the plots of ∆R/R and log-log plot of ∆R/R Vs magnetic field. -

Lecture 19: Review, PN Junctions, Fermi Levels, Forward Bias Context
EECS 105 Spring 2004, Lecture 19 Lecture 19: Review, PN junctions, Fermi levels, forward bias Prof J. S. Smith Department of EECS University of California, Berkeley EECS 105 Spring 2004, Lecture 19 Prof. J. S. Smith Context The first part of this lecture is a review of electrons and holes in silicon: z Fermi levels and Quasi-Fermi levels z Majority and minority carriers z Drift z Diffusion And we will apply these to: z Diode Currents in forward and reverse bias (chapter 6) z BJT (Bipolar Junction Transistors) in the next lecture. Department of EECS University of California, Berkeley 1 EECS 105 Spring 2004, Lecture 19 Prof. J. S. Smith Electrons and Holes z Electrons in silicon can be in a number of different states: Department of EECS University of California, Berkeley EECS 105 Spring 2004, Lecture 19 Prof. J. S. Smith Electrons and Holes z Electrons in silicon can be in a number of different states: Empty states Fermi level ↕ Band gap ↕ In thermal equilibrium, at each location the electrons will fill the Full States states up to a particular level Department of EECS University of California, Berkeley 2 EECS 105 Spring 2004, Lecture 19 Prof. J. S. Smith Fermi function z In thermal equilibrium, the probability of occupancy of any state is given by the Fermi function: 1 F(E) = E−E f 1+ e kT z At the energy E=Ef the probability of occupancy is 1/2. z At high energies, the probability of occupancy approaches zero exponentially z At low energies, the probability of occupancy approaches 1 Department of EECS University of California, Berkeley EECS 105 Spring 2004, Lecture 19 Prof. -
Prezentacja Programu Powerpoint
Nanostructures – density of states Faculty of Physics UW [email protected] Local density of states Gęstość stanów (ogólnie) można zdefiniować jako: 푁 퐸 = 훿 퐸 − 휀푛 푛 Jak widać po scałkowaniu: 퐸2 퐸2 퐸2 න 푁 퐸 푑퐸 = න 훿 퐸 − 휀푛 푑퐸 = න 훿 퐸 − 휀푛 푑퐸 퐸1 퐸1 푛 푛 퐸1 Przykładowo: 1 1 1 2푚 푁1퐷 퐸 = 훿 퐸 − 휀 푘 = න 훿 푘 − 푘′ 2 푑푘 = 2π 퐸′ 푘 휋 퐸 푘 1 1 푚 푁2퐷 퐸 = 훿 퐸 − 휀 푘 = න 훿 푘 − 푘′ 2휋푘 푑푘 = 2π 2 퐸′ 푘 휋ℏ2 푘 3/2 1 1 1 2푚 푁3퐷 퐸 = 훿 퐸 − 휀 푘 = න 훿 푘 − 푘′ 4휋푘2 푑푘 = 퐸 2π 3 퐸′ 푘 2휋2 ℏ2 푘 2017-06-05 2 Electrons statistics in crystals The case of a semiconductor, in which both the electron gas and hole gas are far from the degeneracy: the probability of filling of the electronic states: 퐸 퐸 휉 − 퐺 − 푒 + 퐸퐺 푓 ≈ 푒 2푘퐵푇 푘퐵푇 푘퐵푇 퐸 = + 퐸 퐸푒 푒 2 푒 퐸푐 and of holes 푓ℎ = 1 − 푓푒 퐸 퐸 휉 − 퐺 − ℎ − 2푘 푇 푘 푇 푘 푇 푓ℎ ≈ 푒 퐵 퐵 퐵 ∞ 휋 න 푥푒−푥 푑푥 = 2 0 퐸푣 퐸퐺 Thus: 퐸 = − − 퐸ℎ 퐸ℎ ∗ 3/2 퐸 휉 − 퐸 −휉 2 푚푒푘퐵푇 − 퐺 푐 푛 휉 = 2 푒 2푘퐵푇 ⋅ 푒푘퐵푇 = 푁 푇 푒 푘퐵푇 2휋ℏ2 푐 ∗ 3/2 퐸 휉 − 휉−퐸 푚ℎ푘퐵푇 − 퐺 − 푣 푝 휉 = 2 푒 2푘퐵푇 ⋅ 푒 푘퐵푇 = 푁 푇 푒 푘퐵푇 2휋ℏ2 푣 2017-06-05 3 Electrons statistics in crystals What is the concentration of carriers for T>0? In the thermodynamic equilibrium for an intrinsic semiconductors (półprzewodniki samoistne), the concentration of electrons in the conduction band is equal to the concnetration of holes in the valence band (because they appear only as a result of excitation from the valence band).