Expression of CD13/Aminopeptidase N by Synovial Fibroblasts: Novel Roles in the Pathogenesis of Rheumatoid Arthritis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

Neprilysin Is Required for Angiotensin-(1-7)
Page 1 of 39 Diabetes NEPRILYSIN IS REQUIRED FOR ANGIOTENSIN-(1-7)’S ABILITY TO ENHANCE INSULIN SECRETION VIA ITS PROTEOLYTIC ACTIVITY TO GENERATE ANGIOTENSIN-(1-2) Gurkirat S. Brara, Breanne M. Barrowa, Matthew Watsonb, Ryan Griesbachc, Edwina Chounga, Andrew Welchc, Bela Ruzsicskad, Daniel P. Raleighb, Sakeneh Zraikaa,c aVeterans Affairs Puget Sound Health Care System, Seattle, WA 98108, United States bDepartment of Chemistry, Stony Brook University, Stony Brook, NY 11794, United States cDivision of Metabolism, Endocrinology and Nutrition, Department of Medicine, University of Washington, Seattle, WA 98195, United States dInstitute for Chemical Biology and Drug Discovery, Stony Brook University, Stony Brook, NY 11794, United States Short Title: Angiotensin-(1-7) and insulin secretion Word count: 3997; Figure count: 8 main (plus 3 Online Suppl.); Table count: 1 Online Suppl. Correspondence to: Sakeneh Zraika, PhD 1660 South Columbian Way (151) Seattle, WA, United States Tel: 206-768-5391 / Fax: 206-764-2164 Email: [email protected] 1 Diabetes Publish Ahead of Print, published online May 30, 2017 Diabetes Page 2 of 39 ABSTRACT Recent work has renewed interest in therapies targeting the renin-angiotensin system (RAS) to improve β-cell function in type 2 diabetes. Studies show that generation of angiotensin-(1-7) by angiotensin converting enzyme 2 (ACE2) and its binding to the Mas receptor (MasR) improves glucose homeostasis, partly by enhancing glucose-stimulated insulin secretion (GSIS). Thus, islet ACE2 upregulation is viewed as a desirable therapeutic goal. Here, we show that although endogenous islet ACE2 expression is sparse, its inhibition abrogates angiotensin-(1-7)-mediated GSIS. However, a more widely expressed islet peptidase, neprilysin, degrades angiotensin-(1-7) into several peptides. -

3 Cleavage Products of Notch 2/Site and Myelopoiesis by Dysregulating
ADAM10 Overexpression Shifts Lympho- and Myelopoiesis by Dysregulating Site 2/Site 3 Cleavage Products of Notch This information is current as David R. Gibb, Sheinei J. Saleem, Dae-Joong Kang, Mark of October 4, 2021. A. Subler and Daniel H. Conrad J Immunol 2011; 186:4244-4252; Prepublished online 2 March 2011; doi: 10.4049/jimmunol.1003318 http://www.jimmunol.org/content/186/7/4244 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2011/03/02/jimmunol.100331 Material 8.DC1 http://www.jimmunol.org/ References This article cites 45 articles, 16 of which you can access for free at: http://www.jimmunol.org/content/186/7/4244.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on October 4, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2011 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology ADAM10 Overexpression Shifts Lympho- and Myelopoiesis by Dysregulating Site 2/Site 3 Cleavage Products of Notch David R. -

ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease
International Journal of Molecular Sciences Review ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease Francesca Tosetti 1,* , Massimo Alessio 2, Alessandro Poggi 1,† and Maria Raffaella Zocchi 3,† 1 Molecular Oncology and Angiogenesis Unit, IRCCS Ospedale Policlinico S. Martino Largo R. Benzi 10, 16132 Genoa, Italy; [email protected] 2 Proteome Biochemistry, IRCCS San Raffaele Scientific Institute, 20132 Milan, Italy; [email protected] 3 Division of Immunology, Transplants and Infectious Diseases, IRCCS San Raffaele Scientific Institute, 20132 Milan, Italy; [email protected] * Correspondence: [email protected] † These authors contributed equally to this work as last author. Abstract: Enzymes, once considered static molecular machines acting in defined spatial patterns and sites of action, move to different intra- and extracellular locations, changing their function. This topological regulation revealed a close cross-talk between proteases and signaling events involving post-translational modifications, membrane tyrosine kinase receptors and G-protein coupled recep- tors, motor proteins shuttling cargos in intracellular vesicles, and small-molecule messengers. Here, we highlight recent advances in our knowledge of regulation and function of A Disintegrin And Metalloproteinase (ADAM) endopeptidases at specific subcellular sites, or in multimolecular com- plexes, with a special focus on ADAM10, and tumor necrosis factor-α convertase (TACE/ADAM17), since these two enzymes belong to the same family, share selected substrates and bioactivity. We will discuss some examples of ADAM10 activity modulated by changing partners and subcellular compartmentalization, with the underlying hypothesis that restraining protease activity by spatial Citation: Tosetti, F.; Alessio, M.; segregation is a complex and powerful regulatory tool. -

Cell-Autonomous FLT3L Shedding Via ADAM10 Mediates Conventional Dendritic Cell Development in Mouse Spleen
Cell-autonomous FLT3L shedding via ADAM10 mediates conventional dendritic cell development in mouse spleen Kohei Fujitaa,b,1, Svetoslav Chakarovc,1, Tetsuro Kobayashid, Keiko Sakamotod, Benjamin Voisind, Kaibo Duanc, Taneaki Nakagawaa, Keisuke Horiuchie, Masayuki Amagaib, Florent Ginhouxc, and Keisuke Nagaod,2 aDepartment of Dentistry and Oral Surgery, Keio University School of Medicine, Tokyo 160-8582, Japan; bDepartment of Dermatology, Keio University School of Medicine, Tokyo 160-8582, Japan; cSingapore Immunology Network, Agency for Science, Technology and Research, Biopolis, 138648 Singapore; dDermatology Branch, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD 20892; and eDepartment of Orthopedic Surgery, National Defense Medical College, Tokorozawa 359-8513, Japan Edited by Kenneth M. Murphy, Washington University School of Medicine, St. Louis, MO, and approved June 10, 2019 (received for review November 4, 2018) Conventional dendritic cells (cDCs) derive from bone marrow (BM) intocDC1sorcDC2stakesplaceintheBM(3),andthese precursors that undergo cascades of developmental programs to pre-cDC1s and pre-cDC2s ultimately differentiate into cDC1s terminally differentiate in peripheral tissues. Pre-cDC1s and pre- and cDC2s after migrating to nonlymphoid and lymphoid tissues. + + cDC2s commit in the BM to each differentiate into CD8α /CD103 cDCs are short-lived, and their homeostatic maintenance relies + cDC1s and CD11b cDC2s, respectively. Although both cDCs rely on on constant replenishment from the BM precursors (5). The cy- the cytokine FLT3L during development, mechanisms that ensure tokine Fms-related tyrosine kinase 3 ligand (FLT3L) (12), by cDC accessibility to FLT3L have yet to be elucidated. Here, we gen- signaling through its receptor FLT3 expressed on DC precursors, erated mice that lacked a disintegrin and metalloproteinase (ADAM) is essential during the development of DCs (7, 13). -
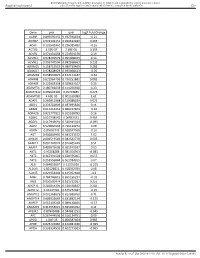
Gene Pval Qval Log2 Fold Change AAMP 0.895690332 0.952598834
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Gene pval qval Log2 Fold Change AAMP 0.895690332 0.952598834 -0.21 ABI3BP 0.002302151 0.020612283 0.465 ACHE 0.103542461 0.296385483 -0.16 ACTG2 2.99E-07 7.68E-05 3.195 ACVR1 0.071431098 0.224504378 0.19 ACVR1C 0.978209579 0.995008423 0.14 ACVRL1 0.006747504 0.042938663 0.235 ADAM15 0.158715519 0.380719469 0.285 ADAM17 0.978208929 0.995008423 -0.05 ADAM28 0.038932876 0.152174187 -0.62 ADAM8 0.622964796 0.790251882 0.085 ADAM9 0.122003358 0.329623107 0.25 ADAMTS1 0.180766659 0.414256926 0.23 ADAMTS12 0.009902195 0.05703885 0.425 ADAMTS8 4.60E-05 0.001169089 1.61 ADAP1 0.269811968 0.519388039 0.075 ADD1 0.233702809 0.487695826 0.11 ADM2 0.012213453 0.066227879 -0.36 ADRA2B 0.822777921 0.915518785 0.16 AEBP1 0.010738542 0.06035531 0.465 AGGF1 0.117946691 0.320915024 -0.095 AGR2 0.529860903 0.736120272 0.08 AGRN 0.85693743 0.928047568 -0.16 AGT 0.006849995 0.043233572 1.02 AHNAK 0.006519543 0.042542779 0.605 AKAP12 0.001747074 0.016405449 0.51 AKAP2 0.409929603 0.665919397 0.05 AKT1 0.95208288 0.985354963 -0.085 AKT2 0.367391504 0.620376005 0.055 AKT3 0.253556844 0.501934205 0.07 ALB 0.064833867 0.21195036 -0.315 ALDOA 0.83128831 0.918352939 0.08 ALOX5 0.029954404 0.125352668 -0.3 AMH 0.784746815 0.895196237 -0.03 ANG 0.050500474 0.181732067 0.255 ANGPT1 0.281853305 0.538528647 0.285 ANGPT2 0.43147281 0.675272487 -0.15 ANGPTL2 0.001368876 0.013688762 0.71 ANGPTL4 0.686032669 0.831882134 -0.175 ANPEP 0.019103243 0.089148466 -0.57 ANXA2P2 0.412553021 0.665966092 0.11 AP1M2 0.87843088 0.944681253 -0.045 APC 0.267444505 0.516134751 0.09 APOD 1.04E-05 0.000587404 0.985 APOE 0.023722987 0.104981036 -0.395 APOH 0.336334555 0.602273505 -0.065 Sundar R, et al. -

Gene Standard Deviation MTOR 0.12553731 PRPF38A
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Gene Standard Deviation MTOR 0.12553731 PRPF38A 0.141472605 EIF2B4 0.154700091 DDX50 0.156333027 SMC3 0.161420017 NFAT5 0.166316903 MAP2K1 0.166585267 KDM1A 0.16904912 RPS6KB1 0.170330192 FCF1 0.170391706 MAP3K7 0.170660513 EIF4E2 0.171572093 TCEB1 0.175363093 CNOT10 0.178975095 SMAD1 0.179164705 NAA15 0.179904998 SETD2 0.180182498 HDAC3 0.183971158 AMMECR1L 0.184195031 CHD4 0.186678211 SF3A3 0.186697697 CNOT4 0.189434633 MTMR14 0.189734199 SMAD4 0.192451524 TLK2 0.192702667 DLG1 0.19336621 COG7 0.193422331 SP1 0.194364189 PPP3R1 0.196430217 ERBB2IP 0.201473001 RAF1 0.206887192 CUL1 0.207514271 VEZF1 0.207579584 SMAD3 0.208159809 TFDP1 0.208834504 VAV2 0.210269344 ADAM17 0.210687138 SMURF2 0.211437666 MRPS5 0.212428684 TMUB2 0.212560675 SRPK2 0.216217428 MAP2K4 0.216345366 VHL 0.219735582 SMURF1 0.221242495 PLCG1 0.221688351 EP300 0.221792349 Sundar R, et al. Gut 2020;0:1–10. doi: 10.1136/gutjnl-2020-320805 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut MGAT5 0.222050228 CDC42 0.2230598 DICER1 0.225358787 RBX1 0.228272533 ZFYVE16 0.22831803 PTEN 0.228595789 PDCD10 0.228799406 NF2 0.23091035 TP53 0.232683696 RB1 0.232729172 TCF20 0.2346075 PPP2CB 0.235117302 AGK 0.235416298 -

Optimal Minimal Panels of Immunohistochemistry for Diagnosis of B-Cell Lymphoma for Application in Countries with Limited Resources and for Triaging Cases Before
AJCP /ORIGINAL ARTICLE Optimal Minimal Panels of Immunohistochemistry for Diagnosis of B-Cell Lymphoma for Application in Countries With Limited Resources and for Triaging Cases Before Referral to Specialist Centers Downloaded from https://academic.oup.com/ajcp/article-abstract/145/5/687/2195691 by World Health Organization user on 09 January 2019 Maria Giulia Disanto, MD,1 Maria Raffaella Ambrosio, MD, PhD,2 Bruno Jim Rocca, MD, PhD,2 Hazem A. H. Ibrahim, FRCPath, PhD,1,3 Lorenzo Leoncini, MD, PhD,2 and Kikkeri N. Naresh, MD, FRCPath1 From the 1Department of Histopathology, Imperial College Healthcare NHS Trust & Imperial College, London, United Kingdom; 2Department of Medical Biotechnologies, Section of Pathology, University of Siena, Siena, Italy; and 3Department of Histopathology, Faculty of Medicine, Mansoura University, Mansoura, Egypt. Key Words: Lymphoma; B-cell lymphoma; Immunohistochemistry; Diagnosis; Classification; Developing countries Am J Clin Pathol May 2016;145:687-695 DOI: 10.1093/AJCP/AQW060 ABSTRACT Lymphomas are a collection of different malignancies “arising” from lymphoid cells. They include about 49 entities, Objectives: Establish and validate optimal minimal and over 19 provisional entities and subsets.1 About 85% of immunohistochemistry panels for usage in a staged lymphomas are of B-cell origin. Precision in lymphoma diag- algorithmic manner for precise diagnosis of B-cell nosis requires expertise and infrastructure. The entities are lymphomas in countries with limited resources. Suggest defined based on morphology, immunohistochemistry (on short panels of immunostains to be used in referring units some occasions in situ hybridization), cytogenetics/fluores- that refer suspected lymphomas to specialist diagnostic cent in situ hybridization (FISH), molecular genetics and clin- centers in resourceful countries. -

CD Markers Are Routinely Used for the Immunophenotyping of Cells
ptglab.com 1 CD MARKER ANTIBODIES www.ptglab.com Introduction The cluster of differentiation (abbreviated as CD) is a protocol used for the identification and investigation of cell surface molecules. So-called CD markers are routinely used for the immunophenotyping of cells. Despite this use, they are not limited to roles in the immune system and perform a variety of roles in cell differentiation, adhesion, migration, blood clotting, gamete fertilization, amino acid transport and apoptosis, among many others. As such, Proteintech’s mini catalog featuring its antibodies targeting CD markers is applicable to a wide range of research disciplines. PRODUCT FOCUS PECAM1 Platelet endothelial cell adhesion of blood vessels – making up a large portion molecule-1 (PECAM1), also known as cluster of its intracellular junctions. PECAM-1 is also CD Number of differentiation 31 (CD31), is a member of present on the surface of hematopoietic the immunoglobulin gene superfamily of cell cells and immune cells including platelets, CD31 adhesion molecules. It is highly expressed monocytes, neutrophils, natural killer cells, on the surface of the endothelium – the thin megakaryocytes and some types of T-cell. Catalog Number layer of endothelial cells lining the interior 11256-1-AP Type Rabbit Polyclonal Applications ELISA, FC, IF, IHC, IP, WB 16 Publications Immunohistochemical of paraffin-embedded Figure 1: Immunofluorescence staining human hepatocirrhosis using PECAM1, CD31 of PECAM1 (11256-1-AP), Alexa 488 goat antibody (11265-1-AP) at a dilution of 1:50 anti-rabbit (green), and smooth muscle KD/KO Validated (40x objective). alpha-actin (red), courtesy of Nicola Smart. PECAM1: Customer Testimonial Nicola Smart, a cardiovascular researcher “As you can see [the immunostaining] is and a group leader at the University of extremely clean and specific [and] displays Oxford, has said of the PECAM1 antibody strong intercellular junction expression, (11265-1-AP) that it “worked beautifully as expected for a cell adhesion molecule.” on every occasion I’ve tried it.” Proteintech thanks Dr. -
HCC and Cancer Mutated Genes Summarized in the Literature Gene Symbol Gene Name References*
HCC and cancer mutated genes summarized in the literature Gene symbol Gene name References* A2M Alpha-2-macroglobulin (4) ABL1 c-abl oncogene 1, receptor tyrosine kinase (4,5,22) ACBD7 Acyl-Coenzyme A binding domain containing 7 (23) ACTL6A Actin-like 6A (4,5) ACTL6B Actin-like 6B (4) ACVR1B Activin A receptor, type IB (21,22) ACVR2A Activin A receptor, type IIA (4,21) ADAM10 ADAM metallopeptidase domain 10 (5) ADAMTS9 ADAM metallopeptidase with thrombospondin type 1 motif, 9 (4) ADCY2 Adenylate cyclase 2 (brain) (26) AJUBA Ajuba LIM protein (21) AKAP9 A kinase (PRKA) anchor protein (yotiao) 9 (4) Akt AKT serine/threonine kinase (28) AKT1 v-akt murine thymoma viral oncogene homolog 1 (5,21,22) AKT2 v-akt murine thymoma viral oncogene homolog 2 (4) ALB Albumin (4) ALK Anaplastic lymphoma receptor tyrosine kinase (22) AMPH Amphiphysin (24) ANK3 Ankyrin 3, node of Ranvier (ankyrin G) (4) ANKRD12 Ankyrin repeat domain 12 (4) ANO1 Anoctamin 1, calcium activated chloride channel (4) APC Adenomatous polyposis coli (4,5,21,22,25,28) APOB Apolipoprotein B [including Ag(x) antigen] (4) AR Androgen receptor (5,21-23) ARAP1 ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 1 (4) ARHGAP35 Rho GTPase activating protein 35 (21) ARID1A AT rich interactive domain 1A (SWI-like) (4,5,21,22,24,25,27,28) ARID1B AT rich interactive domain 1B (SWI1-like) (4,5,22) ARID2 AT rich interactive domain 2 (ARID, RFX-like) (4,5,22,24,25,27,28) ARID4A AT rich interactive domain 4A (RBP1-like) (28) ARID5B AT rich interactive domain 5B (MRF1-like) (21) ASPM Asp (abnormal -

RA0358-C.5-IFU-RUO CD57 / B3GAT1 (Natural Killer Cell
Instructions For Use RA0 35 8-C.5 -IFU -RUO Revision: 1 Rev. Date: Dec. 19, 2014 Page 1 of 2 P.O. Box 3286 - Logan, Utah 84323, U.S.A. - Tel. (800) 729-8350 – Tel. (435) 755-9848 - Fax (435) 755-0015 - www.scytek.com CD57 / B3GAT1 (Natural Killer Cell Marker); Clone NK/804 (Concentrate) Availability/Contents: Item # Volume RA0358-C.5 0.5 ml Description: Species: Mouse Immunogen: Recombinant human B3GAT1 protein Clone: NK/804 Isotype: IgM, kappa Entrez Gene ID: 27087 (Human) Hu Chromosome Loc.: 11q25 Synonyms: 3-Glucuronyltransferase 1; B3GAT1; Galactosylgalactosylxylosylprotein 3-beta- Glucuronosyltransferase 1; GLCATP; GlcUAT-P; Glucuronosyltransferase P; UDP GlcUA Glycoprotein beta 1, 3 Glucuronyltransferase. Mol. Weight of Antigen: ~110kDa Format: Bioreactor Concentrate with 0.05% Azide. Specificity: Anti-CD57 marks a subset of lymphocytes known as natural killer (NK) cells. Anti-CD57 also stains neuroendocrine cells and their derived tumors, including carcinoid tumors and medulloblastoma. Background: Follicular center cell lymphomas often contain many NK cells within the neoplastic follicles. Anti- CD57 can be useful in separating type B3 thymoma from thymic carcinoma when combined with a panel that includes antibodies against GLUT1, CD5, and CEA. Species Reactivity: Human. Does not react with Rat. Others not known. Positive Control: Lymph node or tonsil. Cellular Localization: Cell surface Titer/ Working Dilution: Immunohistochemistry (Frozen and Formalin-fixed): 1:50-1:100 Flow Cytometry: 5-10 µl/million cells Immunofluorescence: 1:50-1:100 Western Blotting: 1:100-1:200 Microbiological State: This product is not sterile. 8° C Storage: 2° C C ScyTek Laboratories, Inc. 205 South 600 West P EmergoEurope (31)(0) 70 345-8570 Logan, UT 84321 Molsnstraat 15 Doc: IFU-Template2-8rev2 U.S.A. -

Flow Cytometry CPT: 88182, 88184, 88185, 88187, 88188, 88189, 86355, 86356, 86357, 86359, 86360, 86361, 86367
Medicare Local Coverage Determination Policy Flow Cytometry CPT: 88182, 88184, 88185, 88187, 88188, 88189, 86355, 86356, 86357, 86359, 86360, 86361, 86367 CMS Policy for Alaska, Arizona, Idaho, Montana, North Dakota, Medically Supportive Oregon, South Dakota, Utah, Washington, and Wyoming ICD Codes are listed Local policies are determined by the performing test location. This is determined by the state on subsequent page(s) in which your performing laboratory resides and where your testing is commonly performed. of this document. Coverage Indications, Limitations, and/or Medical Necessity Flow cytometry (FCM) is a complex process to examine blood, body fluids, CSF, bone marrow, lymph node, tonsil, spleen and other solid tissues. The use of peripheral blood and fine needle aspirate material avoids more invasive procedures for diagnosis. A flow cytometer evaluates the physical and/or chemical characteristics of single cells as the cells pass individually in a fluid stream through a measuring device. Surface receptors, intracellular molecules, and DNA bind with fluorescent dyes that allow detection and evaluation. When light of one wave length excites electrons of certain chemicals to energy levels above their ground state and upon return to ground state emits light of a longer wavelength, fluorescence is produced. A flow cytometer detects cell characteristics by measuring the fluorescence produced by fluorochromes conjugated either directly with cell components or conjugated to antibodies directed against cell components. Indications • Cytopenias and Hypercellular Hematolymphoid Disorders Hematolymphoid neoplasia can present with cytopenias (anemia, leucopenia and/or thrombocytopenia) or elevated leukocyte counts. If medical review and preliminary laboratory testing fails to reveal a cause, bone marrow aspiration and biopsy are indicated to rule out an infiltrative process or a stem cell disorder.