Teva Pharmaceutical Industries
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 | Page Danae Christodoulou, US Food and Drug Administration
Bios Danae Christodoulou, US Food and Drug Administration [email protected] Danae Christodoulou is an Acting Branch Chief in the Office of New Drug Products/OPQ/CDER. Danae joined FDA in 1998 and served as primary reviewer in the Office of New Drugs, as a Chemistry, Manufacturing and Controls Lead and Acting Branch Chief since 2013. Danae has a background in Inorganic Chemistry and received her Ph.D. from the University of Michigan, Ann Arbor, MI. Prior to FDA, Danae worked at Johnson Matthey Inc. R&D Drug Discovery as a Senior Research Chemist and at the National Cancer Institute, in Frederick, MD. Danae served as the Chair for the EI Implementation Working Group at FDA. Marjorie Coppinger, Teva [email protected] Marjorie Coppinger is Head of Global Generic Research Quality (SOD/SSL) for Teva Pharmaceuticals. She received B.A in Chemistry from the University of Notre Dame, Maryland and has over 21 years of experience at Teva Pharmaceuticals in analytical research and development, technical services, manufacturing, quality control, compendial compliance and quality assurance. Currently Ms. Coppinger is Co- Chair of the NJPQCA Compendial Discussion Group and has represented both NJPQCA and GPhA on various USP planning meetings, stakeholder initiatives and project groups for the USP. Elisabeth Corbett, Bristol-Myers Squibb. [email protected] Elisabeth Corbett is an Associate Director, CMC, in Global Regulatory Sciences at Bristol-Myers Squib in Pennington, NJ. She joined Bristol-Myers Squibb as a process engineer in Process Chemistry R&D in 2001. In 2008, she relocated to San Antonio, Texas and served in Quality roles before returning to CMC Regulatory at BMS in 2012. -
Teva to Acquire Cogenesys
TEVA PHARMACEUTICALS AND PERRIGO COMPANY ANNOUNCE THE U.S. LAUNCH OF GENERIC TEMOZOLOMIDE Jerusalem and Allegan, Mich. – August 12, 2013 – Teva Pharmaceutical Industries Ltd. (NYSE: TEVA) and Perrigo Company (NYSE: PRGO;TASE) today announced the launch of the generic equivalent to Temodar® (temozolomide). Teva will manufacture, market and distribute the product in the U.S. and both companies will equally share in the cost and profitability of the product in the U.S. Teva was first to file, making the product eligible for 180 days of marketing exclusivity. This product is the generic equivalent to Temodar® (temozolomide), indicated for the treatment of adult patients with newly diagnosed glioblastoma multiforme concomitantly with radiotherapy and then as maintenance treatment and refractory anaplastic astrocytoma patients who have experienced disease progression on a drug regimen containing nitrosourea and procarbazine. Temodar® had annual sales of approximately $423 million in the United States, according to IMS data as of December 31, 2012. The launch of this product provides a quality alternative, making cancer therapy more cost effective for patients who suffer from this devastating cancer. Perrigo’s Chairman, President and CEO Joseph C. Papa stated, “This first-to-file launch with our partner Teva is another example of our focus to manufacture complex API’s. We are pleased to offer this important product to patients in the United States.” “We are pleased to partner with Perrigo to offer patients a high-quality, less expensive alternative of this important medicine. This launch demonstrates Teva’s commitment to continue to pursue first-to-market opportunities and enhance the value of our portfolio by concentrating on high-margin, low competition markets,” stated Allan Oberman, President and CEO of Teva Americas Generics. -

Teva Respiratory LLC V. Perrigo Pharmaceuticals
Case 1:20-cv-00207 ECF No. 1 filed 03/09/20 PageID.1 Page 1 of 21 IN THE UNITED STATES DISTRICT COURT FOR THE WESTERN DISTRICT OF MICHIGAN ) ) TEVA RESPIRATORY, LLC, and TEVA ) PHARMACEUTICALS USA, INC., ) ) Civil Action No. 20-00207 ) Plaintiffs, ) ) v. ) ) PERRIGO PHARMACEUTICALS CO., and ) PERRIGO COMPANY PLC, ) ) Defendants. ) ) ) COMPLAINT Plaintiffs Teva Respiratory, LLC (“Teva Respiratory”) and Teva Pharmaceuticals USA, Inc. (“Teva USA”) (collectively, “Plaintiffs” or “Teva”), allege against defendants Perrigo Pharmaceuticals Co., and Perrigo Company plc (collectively, “Defendants” or “Perrigo”) as follows: INTRODUCTION 1. This case arises from Perrigo’s deliberate and unlawful attempt to misappropriate Teva’s federally registered trade dress for Teva’s revolutionary ProAir RespiClick® rescue respiratory inhaler and usurp and tarnish Teva’s goodwill. 2. Teva’s ProAir RespiClick® is the only albuterol rescue inhaler on the market that is “breath-activated”; it does not require any hand-breath coordination during inhalation. In other words, Teva’s ProAir RespiClick® enables users to get a rescue dose of albuterol during an asthma or COPD attack simply by breathing in. ProAir RespiClick® is a leading drug for the treatment of bronchospasm. -1- ACTIVE/102581507.4 Case 1:20-cv-00207 ECF No. 1 filed 03/09/20 PageID.2 Page 2 of 21 3. Teva’s ProAir RespiClick® design includes a white body and a distinctive red cap. Teva has a federally registered trademark on the ProAir RespiClick® design. 4. Teva also sells an earlier-generation albuterol rescue inhaler named ProAir® HFA, both as a branded product and as an authorized generic product. -
Bigitech Fact Sheet 2017.12
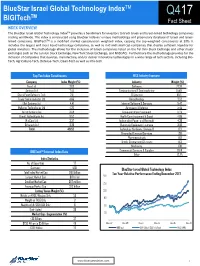
BlueStar Israel Global Technology IndexTM TM Q417 BIGITech Fact Sheet INDEX OVERVIEW The BlueStar Israel Global Technology IndexTM provides a benchmark for investors to track Israeli and Israel-linked technology companies trading worldwide. The index is constructed using BlueStar Indexes’ unique methodology and proprietary database of Israeli and Israel- linked companies. BIGITechTM is a modified market capitalization weighted index, capping the top-weighted constituents at 10%. It includes the largest and most liquid technology companies, as well as mid and small cap companies that display sufficient liquidity for global investors. The methodology allows for the inclusion of Israeli companies listed on the Tel Aviv Stock Exchange and other major exchanges such as the London Stock Exchange, New York Stock Exchange, and NASDAQ. Furthermore the methodology provides for the inclusion of companies that develop, manufacture, and/or deliver innovative technologies in a wide range of tech sectors, including Bio- Tech, Agriculture-Tech, Defense-Tech, Clean-Tech as well as Info-tech. Top Ten Index Constituents GICS Industry Exposure Company Index Weight (%) Industry Weight (%) Nice Ltd 7.63 Software 27.78 Amdocs Ltd 7.44 Semiconductors & Semiconductor 14.49 Check Point Software Tech 7.11 IT Services 10.15 Tower Semiconductor Ltd 4.66 Biotechnology 7.52 Elbit Systems Ltd 4.41 Internet Software & Services 5.47 Mellanox Technologies Ltd 4.26 Aerospace & Defense 5.45 Verint Systems Inc 3.89 Communications Equipment 5.24 Ormat Technologies Inc 3.52 Health Care Equipment & Suppli 4.86 Wix.Com Ltd 3.37 Independent Power and Renewabl 4.36 Orbotech Ltd 3.26 Electronic Equipment, Instrume 3.96 Total 49.52 Technology Hardware, Storage & 1.70 Diversified Financial Services 1.68 Pharmaceuticals 1.49 Hotels, Restaurants & Leisure 1.43 Machinery 1.38 BIGITech™ Internal Index Data Commercial Services & Supplies 0.69 Other 12.35 Index Statistics No. -

Truvada (Emtricitabine / Tenofovir Disoproxil)
Pre-exposure Prophylaxis (2.3) HIGHLIGHTS OF PRESCRIBING INFORMATION These highlights do not include all the information needed to use Recommended dose in HIV-1 uninfected adults: One tablet TRUVADA safely and effectively. See full prescribing information (containing 200 mg/300 mg of emtricitabine and tenofovir for TRUVADA. disoproxil fumarate) once daily taken orally with or without food. (2.3) TRUVADA® (emtricitabine/tenofovir disoproxil fumarate) tablets, for oral use Recommended dose in renally impaired HIV-uninfected Initial U.S. Approval: 2004 individuals: Do not use TRUVADA in HIV-uninfected individuals if CrCl is below 60 mL/min. If a decrease in CrCl is observed in WARNING: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH uninfected individuals while using TRUVADA for PrEP, evaluate STEATOSIS, POST-TREATMENT ACUTE EXACERBATION OF potential causes and re-assess potential risks and benefits of HEPATITIS B, and RISK OF DRUG RESISTANCE WITH USE OF continued use. (2.4) TRUVADA FOR PrEP IN UNDIAGNOSED HIV-1 INFECTION -----------------------DOSAGE FORMS AND STRENGTHS------------------- See full prescribing information for complete boxed warning. Tablets: 200 mg/300 mg, 167 mg/250 mg, 133 mg/200 mg, and 100 Lactic acidosis and severe hepatomegaly with steatosis, mg/150 mg of emtricitabine and tenofovir disoproxil fumarate . (3) including fatal cases, have been reported with the use of nucleoside analogs, including VIREAD, a component of TRUVADA. (5.1) --------------------------------CONTRAINDICATIONS----------------------------- TRUVADA is not approved for the treatment of chronic Do not use TRUVADA for pre-exposure prophylaxis in individuals with hepatitis B virus (HBV) infection. Severe acute unknown or positive HIV-1 status. TRUVADA should be used in exacerbations of hepatitis B have been reported in patients HIV-infected patients only in combination with other antiretroviral coinfected with HIV-1 and HBV who have discontinued agents. -

Which Drugs Are Most Effective for Recurrent Herpes Labialis?
Evidence-based answers from the clinical inquiries Family Physicians Inquiries Network Eiko Tubridy, MD; Gary Kelsberg, MD Valley Family Residency Which drugs are most Program, Renton, Wash Leilani St Anna, MLIS, effective for recurrent AHIP University of Washington Health Sciences Libraries, herpes labialis? Seattle AssistanT EDITOR EvidEncE-basEd answEr Jon O. neher, MD Valley Family Residency daily oral acyclovir or vala- ing to the agent used: valacyclovir reduces Program, Renton, Wash A cyclovir may help prevent her- both healing time and duration of pain, pes simplex labialis (HSL) recurrences famciclovir reduces both in one dosage (strength of recommendation [SOR]: B, form but not another, and acyclovir reduces meta-analysis of randomized controlled only pain duration (SOR: B, single RCTs). trials [RCTs] with heterogeneous results). Several topical medications (acyclovir, No trials compare oral or topical treat- penciclovir, docosanol) modestly decrease ments for HSL outbreaks against each oth- healing time and pain duration—typically er. Oral antivirals modestly reduce healing by less than a day—and require multiple time and duration of pain, varying accord- doses per day (SOR: B, multiple RCTs). Evidence summary The authors of the meta-analysis noted A systematic review and meta-analysis of the that although 9 studies favored the use of an effectiveness of oral and topical nucleoside antiviral drug, only 4 showed statistically sig- antiviral agents to prevent recurrent HSL in nificant differences when compared with pla- immunocompetent people found 11 RCTs cebo, and none of them had a low risk of bias. with a total of 1250 patients that compared They concluded that the review supported us- an active drug against placebo.1 The medi- ing oral acyclovir and valacyclovir to prevent cations were topical 5% acyclovir, topical 1% recurrent HSL.1 penciclovir, and oral acyclovir, valacyclovir, or famciclovir in various doses. -

Wellbutrin Xl : Civil Action Antitrust Litigation : : : No
Case 2:08-cv-02433-GAM Document 419 Filed 05/11/12 Page 1 of 95 IN THE UNITED STATES DISTRICT COURT FOR THE EASTERN DISTRICT OF PENNSYLVANIA IN RE: WELLBUTRIN XL : CIVIL ACTION ANTITRUST LITIGATION : : : NO. 08-2431 (direct) : NO. 08-2433 (indirect) MEMORANDUM McLaughlin, J. May 11, 2012 TABLE OF CONTENTS Page I. Legal and Factual Background.. 2 A. The Drug Approval Process and Regulatory Framework. 2 B. The Citizen Petition Process. 4 C. Wellbutrin IR, Wellbutrin SR, and Wellbutrin XL.. 5 D. Noerr-Pennington Immunity and the Sham Exception. 7 E. Standard of Proof as to Objective Baselessness. 11 II. Overview.. 13 III. Biovail’s Conduct. 15 A. The Anchen Lawsuit. 15 B. The Watson Lawsuit. 26 C. The Abrika Lawsuit. 28 D. The Impax Lawsuit.. 43 E. The Citizen Petition. 50 IV. GSK’s Conduct. 82 A. The Impax and Watson Lawsuits.. 84 B. Biovail’s Citizen Petition. 86 Case 2:08-cv-02433-GAM Document 419 Filed 05/11/12 Page 2 of 95 IN THE UNITED STATES DISTRICT COURT FOR THE EASTERN DISTRICT OF PENNSYLVANIA IN RE: WELLBUTRIN XL : CIVIL ACTION ANTITRUST LITIGATION : : : NO. 08-2431 (direct) : NO. 08-2433 (indirect) MEMORANDUM McLaughlin, J. May 11, 2012 Wellbutrin XL is a once-a-day antidepressant containing the active pharmaceutical ingredient bupropion hydrochloride. It is produced by Biovail Corporation, Biovail Laboratories, Inc., and Biovail Laboratories International SRL1 (together, “Biovail”), and distributed by SmithKline Beecham Corporation and GlaxoSmithKline, PLC (together, “GSK”). The plaintiffs, direct and indirect purchasers of Wellbutrin XL, have sued Biovail and GSK for illegally conspiring to prevent generic versions of Wellbutrin XL from entering the American market by filing sham patent infringement lawsuits and a citizen petition with the Food and Drug Administration (“FDA”), and entering into agreements with generic companies to settle the lawsuits. -

Ormat Technologies Inc
ORMAT TECHNOLOGIES INC MEETING DATE Wed, 06 Nov 2019 13:00 TYPE EGM ISSUE DATE Fri, 25 Oct 2019 MEETING LOCATION 6140 Plumas Street, Reno, NV 89519 CURRENT INDICES PIRC Global SECTOR Electric services COMPANY OVERVIEW Ormat Technologies is a provider of renewable power and energy technology. The Company is based in Reno, Nevada, and has over 150 power plants. MEETING SPECIFIC INFORMATION Ordinary resolutions are approved by simple majority of the votes cast. PROPOSALS ADVICE 1 Board Proposal to Declassify the Board For It is considered that staggered elections do not pursue shareholders’ best interest, as they entrench the board against hostile takeovers. In this sense, the Board’s proposal is welcomed as it will introduce annual election for all directors of the board, which is considered to be best practice. 2 Adjourn Meeting Oppose The Board requests authority to adjourn the special meeting until a later date or dates, if necessary, in order to permit further solicitation of proxies if there are not sufficient votes at the time of the special meeting to approve the merger. An oppose vote is recommended to any adjournment or postponement of meetings if a sufficient number of votes are present to constitute a quorum. It is considered that where a quorum is present, the vote outcome should be considered representative of shareholder opinion. ORMAT TECHNOLOGIES INC 06 Nov 2019 EGM 1 of 5 BOARD AND COMMITTEE COMPOSITION (post-Meeting) INDEPENDENT BY DIRECTOR GENDER PIRC COMPANY BOARD AC RC NC SC TENURE Dan Falk M No Yes NED C M M - 14 Todd C. -

Reference ID: 2998411
• New onset or worsening renal impairment: Can include acute HIGHLIGHTS OF PRESCRIBING INFORMATION renal failure and Fanconi syndrome. Assess creatinine clearance These highlights do not include all the information needed to use (CrCl) before initiating treatment with COMPLERA. Monitor CrCl COMPLERA safely and effectively. See full prescribing and serum phosphorus in patients at risk. Avoid administering information for COMPLERA. COMPLERA with concurrent or recent use of nephrotoxic drugs. (5.3) COMPLERATM (emtricitabine/rilpivirine/tenofovir disoproxil • Caution should be given to prescribing COMPLERA with drugs fumarate) tablets that may reduce the exposure of rilpivirine. (5.4) Initial U.S. Approval: 2011 • Caution should be given to prescribing COMPLERA with drugs with a known risk of Torsade de Pointes. (5.4) WARNINGS: LACTIC ACIDOSIS/SEVERE HEPATOMEGALY WITH STEATOSIS and POST TREATMENT ACUTE • Depressive disorders: Severe depressive disorders (depressed EXACERBATION OF HEPATITIS B mood, depression, dysphoria, major depression, mood altered, negative thoughts, suicide attempt, suicidal ideation) have been See full prescribing information for complete boxed warning. reported. Immediate medical evaluation is recommended for severe depressive disorders. (5.5) • Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of • Decreases in bone mineral density (BMD): Consider monitoring nucleoside analogs, including tenofovir disoproxil BMD in patients with a history of pathologic fracture -

2020 Annual Report Products
Products 2020 Annual Report From the CEO Dear Fellow Shareholders, Perrigo’s transformation to a pure-play Consumer Self-Care Company has come a long way LQMXVWWZRVKRUW\HDUV:HKDYHUHVWRUHGVXVWDLQDEOHWRSOLQHJURZWKGHOLYHUHGRQRXU¿QDQFLDO SURPLVHVUHFRQ¿JXUHGRXUSRUWIROLRRIEXVLQHVVHVXSGDWHGWKH,7LQIUDVWUXFWXUHDQGSURFHVVHV of the Company, expanded capacity, upgraded leadership talent, installed business intelligence capabilities, built a new product pipeline of over $500 million and re-instilled a sense of pride and energy among our 11,000 team members. Making this even more remarkable, is that we kept the WUDQVIRUPDWLRQRQWUDFNLQWKHIDFHRIWKHJOREDO&29,'SDQGHPLF,KRSH\RXDUHDVSURXGRI3HUULJR¶VJOREDOWHDP DV,DPIRUKRZWKH\ZRUNHGWRNHHSHDFKRWKHUVDIHNHSWRXUHVVHQWLDOSURGXFWVÀRZLQJDQGNHSWRXUWUDQVIRUPDWLRQ to a consumer self-care company on track through all of the personal and professional uncertainty that came their way LQ7KH\DUHKHURHV As a result of their efforts, Perrigo delivered strong net sales growth for the second year in a row in 2020 and World-wide Consumer sales reached a new record high. Equally important, the team stabilized adjusted operating income after a few years of decline even as we invested over $50 million in our business and overcame $35 million RIXQSODQQHGKHDGZLQGVGXHSULPDULO\WR&29,'UHODWHGVDIHW\FRVWVDQGEXVLQHVVLPSDFWIURPWKHZHDNFROG FRXJKDQGÀXVHDVRQUHODWHGWR&29,'¶VLPSDFWRQSXEOLFOLIH$OOLQDOOZHKDGDYHU\VWURQJ\HDU Our transformation efforts reached an essential milestone after the year closed when we announced the sale of RXU3UHVFULSWLRQ3KDUPDFHXWLFDOVEXVLQHVVWR$OWDULV&DSLWDO3DUWQHUV//&7KHWUDQVDFWLRQUHLQIRUFHVRXUDELOLW\ -

Caesarea Forum - Xvii
CAESAREA FORUM - XVII PROGRAM Chair: Dr. Yuval Steinitz, Minister of Finance Academic Director of the Forum: Mr. David Brodet Wednesday and Thursday, 1st -2nd July 2009 Hotel Royal Beach, Eilat WEDNESDAY, 1st JULY 2009 8:30 - 9:30 Registration and assembly 9:30 - 13:00 First Session : "The Requisite Size of the Government - between Economy and Politics" [The Olive and Acacia Hall] Presentation of the Team's Recommendations: Prof. Joseph Zeira , Department of Economics, The Hebrew University of Jerusalem Presenters: Prof. Avishai Braverman, Minister of Minority Affairs Mr. Yoram Ariav , Director General and Acting Head of the Budget Department, Ministry of Finance Prof. Zvi Eckstein , Deputy Governor, Bank of Israel Dr. Michel Strawczynski, Director, Macro-Economic and Policy Department, Bank of Israel Prof. Omer Moav, Department of Economics, The Hebrew University of Jerusalem Prof. Manuel Trajtenberg , Head, National Economic Council, Prime Minister's Office Ms. Yael Andorn, Director General, Amitim Pension Funds Dr. Yossi Bachar , Economic Consulting and Business Development Ltd. Dr. Eldad Shidlovsky , Head, Economics and Research Department, Ministry of Finance Plenary discussion 13:00 - 14:15 Minister of Finance's Session : [The Ranch House Restaurant] Luncheon Address by the Dr. Yuval Steinitz, Minister of Finance 14:30 - 18:15 Second Session : "The Future of Growth Promotion in Israel: A Return to Boosting Avant-garde Industries and Scientific Technological Innovation" [The Olive and Acacia Hall] 14:30-16:30 Presentation of the Team's Recommendations: Prof. Arnon Bentor, Dean, Civil and Environmental Engineering Faculty, and Senior Researcher, The Neaman Institute, The Technion Presenters: Mr. Eli Hurvitz, Chairman, Teva Pharmaceutical Industries Ltd. -

Comparable Efficacy with Entecavir Monotherapy and Tenofovir
BMJ Open Gastroenterol: first published as 10.1136/bmjgast-2015-000030 on 31 December 2015. Downloaded from Hepatology Comparable efficacy with entecavir monotherapy and tenofovir–entecavir combination in chronic hepatitis B patients Sumbella Baqai,1 James Proudfoot,2 Ronghui Xu,3,4 Steve Kane,5 Margaret Clark,6 Robert Gish,7,8,9,10,11 To cite: Baqai S, Proudfoot J, ABSTRACT et al Summary box Xu R, . Comparable Objectives: The long-term goal for chronic hepatitis B efficacy with entecavir patients is to maintain viral suppression in order to monotherapy and tenofovir– What is already known about this subject? reduce disease progression risk. Because patients with entecavir combination in ▸ Finding effective therapy for patients with chronic hepatitis B patients. previous treatment failure may have multiple viral chronic hepatitis B virus (HBV) infection who BMJ Open Gastro 2015;2: resistance mutations, finding effective therapy is e000030. doi:10.1136/ challenging. Because recent studies have shown that have had previous treatment failure, many of bmjgast-2015-000030 the combination of entecavir and tenofovir is effective whom have multiple viral resistance mutations, in achieving virological response in many patients with is a challenge. prior treatment failure and multiple drug resistance ▸ Several recent studies have shown that combin- ation therapy with entecavir (ETV) and tenofovir Received 27 January 2015 mutations, we compared outcomes with this Revised 22 April 2015 combination versus monotherapy. (TDF) is effective in achieving virological by copyright. Accepted 30 April 2015 Methods: With a retrospective chart review we response in the majority of patients with prior compared results in 35 patients with previous treatment treatment failure and multiple drug resistance failure treated with the entecavir-tenofovir combination to mutations.